
Comparisons between gelatin-tussah silk fibroin/hydroxyapatite and gelatin-Bombyx mori silk fibroin/hydroxyapatite nano-composites for bone tissue engineering
Abstract
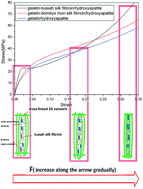
In this research, gelatin-tussah silk fibroin/hydroxyapatite (GEL-TSF/HAp), gelatin-Bombyx mori silk fibroin/hydroxyapatite (GEL-BMSF/HAp), and gelatin/hydroxyapatite (GEL/HAp) nano-composites were synthesized by a novel in situ precipitation method. Characterizations, including surface morphology, elemental composition and distribution, structure of the crystalline phase, mechanical strength, thermal stability, and in vitro cytocompatibility, were carried out. Investigations on the crystalline phase showed that rod-like HAp crystallites in the GEL-TSF/HAp composite had a higher aspect ratio than those in the GEL-BMSF/HAp composite and the GEL/HAp composite. In addition, the GEL-TSF/HAp composite also presented better thermal stability than the other two composites revealed by differential thermal analysis (DTA) and thermogravimetric analysis (TGA). Mechanical properties testing indicated that the GEL-TSF/HAp composite had a higher elastic modulus at low strain and higher compressive modulus at high strain simultaneously than the other two composites. An in vitro cell culture showed that MG63 osteoblast-like cells on the GEL-TSF/HAp membrane took on higher proliferative potential than those on the GEL-BMSF/HAp membrane. These results indicated that compared to the GEL-BMSF/HAp composite, the GEL-TSF/HAp composite was more suitable for bone tissue engineering (BTE) applications.
 Please wait while we load your content...
Please wait while we load your content...

