DOI:
10.1039/C5RA14197D
(Paper)
RSC Adv., 2015,
5, 65575-65582
Microwave-assisted synthesis of pyrite FeS2 microspheres with strong absorption performance
Received
18th July 2015
, Accepted 28th July 2015
First published on 28th July 2015
Abstract
Pyrite FeS2 microspheres with an average size of approximately 1.1 μm were successfully synthesised in high yield via a facile and efficient microwave-assisted method. X-ray diffraction patterns and high-resolution transmission electron micrographs showed that the microspheres had a pure single crystalline phase. Scanning electron micrographs and transmission electron micrographs illustrated that the microspheres were uniform and well-distributed. The absorption spectra of the as-obtained microspheres showed that pyrite exhibited high absorbance in the visible region. The surfactants influenced the phase, morphology and absorption property of the products. Long reaction time allowed the formation of uniform pyrite FeS2 microspheres with smooth surfaces. The microwave-assisted method yielded products with better morphology and absorption properties than the high-pressure solvothermal method.
1. Introduction
Also known as “fool's gold”, iron pyrite (FeS2) has attracted considerable interest as a promising photovoltaic application material because of its suitable band gap (Eg = 0.95 eV), high light absorption coefficient (α > 105 cm−1 for hμ > 1.3 eV),1 low cost, nontoxicity and vast abundance when compared with other candidates. The optical absorption coefficient of pyrite FeS2 is considerably higher than that of crystalline silicone, making pyrite FeS2 applicable as an absorbent for thin-film solar cells and photovoltaic cells.2 However, the coexistence of impurity phases (FeS, Fe3S4 and marcasite FeS2) in natural bulk pyrite FeS2, complicates the procurement of pure-phase pyrite FeS2, these impurities result in low-efficiency photovoltaic conversion,3,4 and limit the development of relevant devices. Therefore, new methods to synthesise pure-phase, homogeneous pyrite FeS2 must be developed.
To date, various routes for pyrite FeS2 synthesis have been reported. Xia et al.5 utilised a one-step hydrothermal route and successfully obtained FeS2 nanocrystals with diameters ranging from 10 nm to 35 nm, Zhang et al.6 synthesised bud-like FeS2 microspheres via a solvothermal method. Sulphurising Fe2O3 nanorods,7 chemical vapour deposition,8 solvent-induced oriented attachment9 and hot injection route10 have also been used to obtain pyrite FeS2. In addition, pyrite has been synthesised in different shapes, including micro-octahedral,11 nanowires,12 nanocubes13 and nanotubes.14 However, conventional methods are usually disadvantaged by long reaction times, uneven heating, low-yield, and complex processes. Thus, exploring a simple and efficient method to synthesise pure-phase FeS2 remains a challenge.
Microwave-assisted methods have gradually developed in recent years, and they are now applied in many areas, such as organic and inorganic syntheses. The interaction between the absorbing medium and microwaves in microwave-assisted methods generates heat and allows rapid and homogeneous heating. Different from conventional heating methods, microwave-assisted methods facilitate homogeneous, rapid, facile, clean and easy-to-control heating.15,16 Recent studies have used microwave-assisted methods to synthesise Ag2S,17 NiS2,18 CuS2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 and Fe3O4.20 Kim EJ et al.21 employed a microwave-assisted method to obtain pyrite FeS2, however, their use of a glove box containing nitrogen gas added inconvenience and complexity to the synthesis process. Similar experiments obtained products with inhomogeneous morphology and large size.22,23 Considering the aforementioned problems, this study utilized a simple and efficient microwave-assisted system to synthesise pure-phase, uniform and regularly shaped pyrite FeS2. The temperature detector of the synthesis system was placed in the interior of the solution to realize accurate temperature control. Stirring throughout the heating process ensured completion of the reaction. Pure-phase, uniform-morphology and highly crystalline pyrite FeS2 microspheres with an average size of approximately 1.1 μm were obtained. The influences of surfactants and reaction time on the phase and morphology of the products were investigated.
19 and Fe3O4.20 Kim EJ et al.21 employed a microwave-assisted method to obtain pyrite FeS2, however, their use of a glove box containing nitrogen gas added inconvenience and complexity to the synthesis process. Similar experiments obtained products with inhomogeneous morphology and large size.22,23 Considering the aforementioned problems, this study utilized a simple and efficient microwave-assisted system to synthesise pure-phase, uniform and regularly shaped pyrite FeS2. The temperature detector of the synthesis system was placed in the interior of the solution to realize accurate temperature control. Stirring throughout the heating process ensured completion of the reaction. Pure-phase, uniform-morphology and highly crystalline pyrite FeS2 microspheres with an average size of approximately 1.1 μm were obtained. The influences of surfactants and reaction time on the phase and morphology of the products were investigated.
2. Experimental section
2.1 Materials and equipment
Anhydrous FeCl3, polyvinyl pyrrolidone (PVP K-30, Mw = 40000), Na2S, sulphur powder and ethylene glycol (EG) were of analytical purity and used without further purification. PVP was purchased from Sigma-Aldrich Co. LLC. All other reagents used in the experiments were procured from Aladdin Co. Ltd. Reactions were performed in a customized 100 mL Teflon-lined chamber, and heating was conducted in a microwave hydrothermal synthesis system (Beijing Xianghu, XH-8000).
2.2 Synthesis of pyrite FeS2
In a typical synthesis, 2 mmol anhydrous FeCl3 and 0.6 g PVP were dissolved in 30 mL EG under vigorous and continuous magnetic stirring. Then, 2 mmol Na2S was added to the solution, in which 4 mmol sulphur powder was dispersed via ultrasonication for approximately 30 min. The solution was transferred to a Teflon-lined chamber and equipped on the microwave reactor. The temperature was increased to 200 °C, and the reaction was conducted for 1.5 h via microwave irradiation. After the reaction, the black products were cooled to room temperature naturally and then collected via centrifugation at 8500 rpm. The products were sequentially washed with distilled water, carbon disulphide and absolute ethanol for several times. The final products were adequately dried in a vacuum oven at 60 °C for 5 h.
2.3 Characterisation
The crystal structures of the products were investigated using a Bruker AXS XRD-D8 Focus X-ray diffractometer equipped with graphite monochromatised Cu Kα radiation (λ = 0.15406 nm). An FEI Quanta 200F scanning electron microscope was used to characterize the morphology of the as-prepared products, and energy dispersive X-ray (EDX) spectroscopy was performed using scanning electron microscopy (SEM). High-resolution transmission electron micrographs were recorded on an FEI F20 field-emission transmission electron microscope at an acceleration voltage of 200 kV. The Raman spectra were obtained on a Horiba Jobin Yvon LabRAM HR800 Raman spectrometer operating with a 512 nm laser. Absorption properties were acquired by using a HITACHI U-4100 UV-Vis-NIR spectrophotometer. X-ray photoelectron spectroscopy (XPS) was performed on a Thermo ESCALAB 250XI electron spectrometer using monochromatised Al Kα radiation.
3. Results and discussion
3.1 Characterisation and optical property of pyrite FeS2 microspheres
The XRD pattern of the pyrite FeS2 products is shown in Fig. 1. All diffraction peaks of the as-obtained samples were readily and perfectly indexed to the Standard Card No. 42-1340. No apparent impurity peaks (such as from marcasite FeS2 and FeS) appeared, revealing the formation of high-purity pyrite FeS2. The sharp and narrow peaks in the XRD pattern indicated the excellent crystallinity of the as-obtained pyrite FeS2 products. The SEM images of the as-obtained pyrite FeS2 microspheres are shown in Fig. 2a and b, these microspheres showed regular shapes, good uniformity and smooth surfaces. The corresponding pyrite size distribution of Fig. 2b is shown in Fig. 2c, where the sizes of the products ranged from 600 nm to 1.2 μm. The histogram shows that the samples were mostly approximately 1.1 μm in size. The chemical composition of the as-obtained pyrite FeS2 microspheres was characterized by EDX analysis. As shown in Fig. 2d, the products had an iron-to-sulphur ratio of approximately 1:2.2, which is consistent with FeS2. The Cu peaks in the image were derived from the conductive Cu substrate used to prepare the samples. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) were performed to study further the morphology and structure of the samples. The typical TEM image in Fig. 2e shows that the samples were well dispersed. In addition, the fringe spacings in the HRTEM image in Fig. 2f were 0.24 and 0.36 nm, which matched the lattice spacings of the (210) and (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 22) planes of bulk pyrite FeS2, respectively.
22) planes of bulk pyrite FeS2, respectively.
 |
| | Fig. 1 XRD pattern of the pyrite FeS2 microspheres and JCPDS Card No. 42-1340. | |
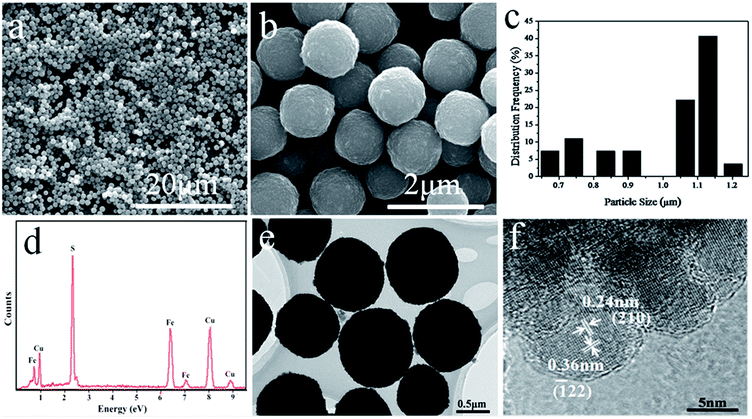 |
| | Fig. 2 (a)–(b) SEM images of the pyrite FeS2 microspheres. (c) Particle size distribution corresponding to the image b. (d) EDX spectra of the samples. (e)–(f) TEM and HRTEM images of the pyrite FeS2 microspheres. | |
Raman spectroscopy has lower impurity detection limit than XRD and can differentiate FeS2 phases (FeS, Fe3S4 and FeS2).24 Thus, Raman spectroscopy was applied in the present study to further confirm the pure phase of pyrite FeS2 microspheres. As shown in Fig. 3a, sharp peaks at 342, 377 and 429 cm−1 were the characteristic active modes of pyrite FeS2 corresponding to S2 liberation (Eg), S–S in-phase stretch (Ag) and coupled liberation (Tg) modes, respectively.25 The high phase purity of the products was confirmed by the absence of the Raman peaks of other phases such as marcasite FeS2, FeS and Fe3S4. Fig. 3b and c show the XPS spectra of the pyrite FeS2 microspheres. In Fig. 3b, the peaks at 707.1 and 719.9 eV corresponded to Fe 2p3/2 and Fe 2p1/2 binding energies, respectively, which were characteristics of pyrite FeS2.26,27 The S 2p3/2 and S 2p1/2 peaks at 162.4 and 163.6 eV (Fig. 3c), respectively, were also consistent with the sulphur binding energy of previously reported pyrite FeS2.26,27 These characterization results further confirmed the pure phase and high quality of the as-obtained pyrite FeS2 microspheres. Pyrite is a promising material for solar cells mainly because of its favourable optical properties. As shown in Fig. 4a, the absorption spectral intensity of the pyrite samples decreased with increasing wavelength. The samples exhibited a broad optical absorption from the ultraviolet to near-infrared (NIR) region, and the high absorbance in the visible region indicated the potential of the product for solar energy absorption. The band gap of the samples was obtained by using the extrapolated data from the absorption spectrum. Basing from Fig. 4b, we calculated the band gap of the samples to be approximately 1.02 eV. This value is close to the 0.95 eV in a previous study.28
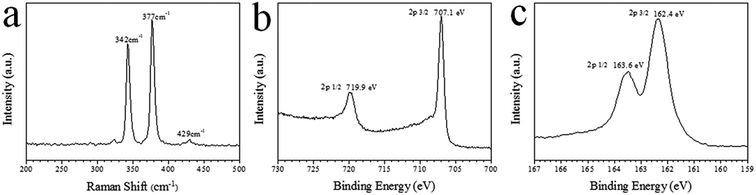 |
| | Fig. 3 (a) Raman spectra of the pyrite FeS2 microspheres. (b)–(c) XPS analysis of the pyrite microspheres: (b) Fe region and (c) S region. | |
 |
| | Fig. 4 UV-Vis-NIR absorption spectra and band gap plot of pyrite FeS2 microspheres. | |
3.2 Effect of surfactants on the phase, morphology and optical property of products
In this study, PVP acted as a typical surfactant. The influence of other surfactants was investigated to improve the quality of pyrite FeS2 products. As common surfactants in synthetic reactions, hexadecyl trimethyl ammonium bromide (CTAB, 0.6 g) and trioctyl-phosphine oxide (TOPO, 0.6 g) were used as controls in this study. Fig. 5 shows the XRD patterns of the products obtained with different surfactants. Pure-phase pyrite FeS2 was obtained when no surfactant was added in the reaction system. Meanwhile, marcasite FeS2 appeared along with pyrite FeS2 when CTAB was used as the surfactant. The condition when TOPO served as the surfactant was similar to that when CTAB was used, and the phases of the products were both pyrite FeS2 and marcasite FeS2. However, the relative peak intensities of marcasite FeS2 were slightly stronger while the peak intensities of pyrite FeS2 became weaker when TOPO rather than CTAB was the surfactant. This result indicates that the two phases of the two product groups had different relative contents. The XRD patterns showed that CTAB and TOPO acted as phase-transfer catalysts in the experiment. The chemical reactions in FeS2 synthesis can be explained by the following reactions:| | |
2Fe3+ + S2− → 2Fe2+ + S
| (1) |
| | |
2CH2OH–CH2OH + S → CH3COCOCH3 + S2− + 2H+ + 2H2O
| (2) |
| | |
(n − 1)S + S2− → Sn2−
| (3) |
| | |
Fe2+ + Sn2− + S2− → FeS2 + Sn−12−
| (4) |
 |
| | Fig. 5 XRD patterns of the products obtained with different surfactants. | |
Fe3+ is reduced to Fe2+ upon adding the reagents into the solution [eqn (1)]. Then, sulphur powder is reduced to S2− at high temperatures with EG as the reducing agent [eqn (2)].29,30 The unreduced elemental S reacts with S2− to form polysulphides [eqn (3)].23 Afterward, FeS2 is generated by the reaction between polysulphides and Fe2+, [eqn (4)].21 The formation mechanism by which FeS2 crystallises into a pyrite or marcasite structure at low temperatures (<300 °C) remains unclear to date. The generation of pyrite and marcasite may be caused by electrostatic interactions between polysulphide species and pyrite or marcasite growth surfaces.31 The protonated ends of polysulphides favoured marcasite growth sites, whereas negative polysulphide ions were attracted to the positive pyrite growth sites. Marcasite formed when polysulphide ends were protonated. The ionisation of the cationic surfactant CTAB promoted the protonation of the polysulphide ends and thus resulted in the formation of marcasite. Recent research has revealed that a relatively high concentration of intermediate sulphur species results in marcasite formation.32 Moreover, the addition of TOPO increases the solubility of materials and promotes the stability of FeS2.33–35 The consequent increase in the reduction rate of sulphur powder and the constant high concentration of the intermediate sulphur species led to the appearance of marcasite.
The corresponding SEM images of the products using different surfactants are shown in Fig. 6. Products with irregular shapes agglomerated when no surfactant was added in the reaction system. Some of the products displayed the morphology of two sheets inserting into each other, when CTAB was used as the surfactant (inset of Fig. 6b). Meanwhile, the products appeared as aggregates of many small sheets without apparent regular shape when TOPO served as the surfactant. Finally, uniform and regularly shaped microspheres with relatively smooth surfaces were obtained when PVP was the surfactant. Given its long molecular chains, PVP is a good capping agent and can be adsorbed and covered on particle surfaces.36–38 The steric effect of PVP prevented particle agglomeration allowed the formation of well dispersed products. Previous studies reported that the preferential adsorption of surfactant molecules on different crystal facets is vital to direct the growth of particles into different morphologies by controlling the growth rates along different crystal axes.39–42 Different surfactants exhibiting varying structures, solubilities, charges and polarities greatly affect crystal growth by interacting with different facets.43 Thus, the employment of CTAB and TOPO as surfactants in the present study can yield products with different shapes. In conclusion, PVP favoured the best morphology of the products among the tested surfactants.
 |
| | Fig. 6 SEM images of the products obtained (a) without and with different surfactants (b: CTAB, c: TOPO, d: PVP). | |
The absorption spectra of the products obtained with CTAB and TOPO as surfactants are shown in Fig. 7. The curves of the two products had similar profiles and their absorption intensities decreased with increasing wavelength. The absorbance values of the products obtained using CTAB and TOPO as surfactants were lower than that of the products obtained using PVP as the surfactant. Given its small energy band gap of 0.34 eV, marcasite is unsuitable for photoelectric translation, the existence of marcasite affects the absorption property of pyrite.44 Morphology, size, shape and uniformity also greatly influence the absorption performance of products.10,45–48 The products obtained using PVP as the surfactant showed regular shapes, improved uniformity and small sizes. Besides, it's reported that facets may affect the absorption behavior of products,49 results could be influenced since the spherical pyrite products were unfaceted, whereas the irregularly shaped products obtained using CTAB and TOPO as surfactants were terminated by different facets. The synthetic effects of the above mentioned factors finally led to the difference in absorbance of the products. The microspheres obtained with PVP as the surfactant exhibited the best absorption property among the products. In addition, the products obtained with CTAB as the surfactant showed slightly higher absorbance in the visible region than the products obtained with TOPO as the surfactant. This result may be attributed to the relatively higher proportion of pyrite when CTAB used as surfactant. Among all the products, those obtained with PVP as the surfactant showed the best absorption property.
 |
| | Fig. 7 Absorption spectra of the products obtained with different surfactants. | |
3.3 Formation of pyrite FeS2 microspheres
Reaction-time dependence experiments were conducted to reveal the formation of pyrite microspheres and to explore the effect of reaction time on the phases and morphologies of the products. Fig. 8 displays the SEM images of the products obtained for different reaction periods of 0.5, 1 and 1.5 h while maintaining other conditions constant. When the reaction time was 0.5 h, massive small microspheres were altered, but they were inhomogeneous and considerably differed in size. Meanwhile, many small particles appeared on the microsphere surfaces. After 30 min of microwave irradiation, the microspheres became uniform and regular, and their surfaces became smooth. When the reaction time was prolonged to 1.5 h, the microspheres dispersed homogeneously, and their surfaces became increasingly smooth. The microspheres slightly increased in size as the reaction time was further prolonged. The corresponding XRD patterns of the products prepared at different reaction times are shown in Fig. 9. Pyrite FeS2 was produced in all three reaction periods. The products had similar XRD patterns but relative peak intensity levels. In addition, the diffraction peaks became stronger as the reaction time was prolonged, this finding can be attributed to the improvement of product crystallinity. Thus, the formation of pyrite FeS2 microspheres may be similar to that in previous reports.50–52 In particular, the products nucleated rapidly under microwave irradiation. Different nucleation speeds and growth rates resulted in the formation of large microspheres and small particles. As the reaction time was prolonged, the particles aggregated and continued to grow, and the products consequently increased in size. As the dissolved quantity of PVP was increased, further aggregation and growth of microspheres were prevented by the steric effect of PVP. Ultimately, the dispersion of the products improved.
 |
| | Fig. 8 SEM images of the as synthesized pyrite FeS2 with different reaction times (a: 0.5, b: 1, c: 1.5 h). | |
 |
| | Fig. 9 XRD patterns of as synthesized pyrite FeS2 at different reaction times. | |
3.4 Comparison between microwave-assisted heating and high-pressure solvothermal heating method
Microwave-assisted method exhibits many advantages over conventional heating methods. A control experiment with the high-pressure solvothermal reaction was conducted to compare the method. The reagents of the solvothermal experiment were completely same to those of the microwave-assisted experiment, and the reaction was conducted at 200 °C for 1.5, 12 and 24 h, respectively. The XRD patterns of the products obtained at different reaction times are shown in Fig. 10. When the reaction time was 1.5 h, the products were mostly sulphur, and low intensity peaks of pyrite FeS2 were observed. This result indicates that an incomplete reaction occurred. As the reaction time was prolonged to 12 and 24 h, the products were both pure-phase pyrite FeS2. Fig. 11 shows the SEM images of the products obtained at different reaction times. Many irregularly shaped particles formed after 1.5 h of reaction, EDX shows that the elements in the particles were sulphur and iron, and the sulphur-to-iron ratio was 88.8:11.2 (inset of Fig. 11a). This result is consistent with the XRD pattern. When the reaction time was extended to 12 h, some small microspheres with rough surfaces formed, but the products were inhomogeneous dispersed and non-uniform. Even small irregular particles were found at this reaction period. The products after 24 h of reaction became uniform, but their surfaces remained rough with some agglomerations. In addition, the pyrite particles obtained using the solvothermal method were considerably larger than those obtained using the microwave-assisted method. These size differences and agglomeration might be attributed to the uneven heating in the high-pressure solvothermal method. In this technique, heat is generated from the oven and is transferred from the outside of chamber to the solution, causing an uneven heating environment. This heating condition diversifies the nucleation and crystal growth, and thus triggers the formation of non-uniform products. In addition, the incomplete reaction in the high-pressure solvothermal method for 1.5 h indicated that the heating rate of the solvothermal method was considerably lower than that of the microwave-assisted method. This result indirectly indicates the superiority of the microwave-assisted method used in this work over the solvothermal method.
 |
| | Fig. 10 XRD patterns of products obtained using the high-pressure solvothermal method at different reaction times. | |
 |
| | Fig. 11 SEM images of products obtained using the high-pressure solvothermal method at different reaction times. | |
The absorption spectra of the products obtained using the solvothermal method at different reaction times are shown in Fig. 12. When the reaction time was 1.5 h, the absorbance of the products was very low, especially at the Vis-NIR region. This finding resulted from the low amount of pyrite FeS2 because the majority of the products obtained in this condition were sulphur. When the reaction time was prolonged to 12 h, the absorbance of the products considerably increased, and the products exhibited broad absorption from the ultraviolet to NIR region. Meanwhile, a similar curve with higher absorbance was obtained when the reaction time was 24 h. All products were pyrite FeS2, the increased amount of pyrite increased the absorbance of the products. The products of the solvothermal method increased in absorbance with reaction time, but this absorbance remained lower than that of the products obtained using the microwave-assisted method. This result might be attributed to the effects of the size, shape, crystallization and uniformity of the products on their absorption performance.10,45–49 Moreover, the small spherical products obtained using the microwave-assisted method would increase the surface area of pyrite FeS2 and improve light harvesting.22,53 Therefore, the microwave-assisted heating method accelerated the synthesis of uniform pyrite FeS2 and the absorption property of the products better than the high-pressure solvothermal heating method.
 |
| | Fig. 12 Absorption spectra of products obtained using the high-pressure solvothermal method at different reaction times. | |
4. Conclusions
Pure-phase pyrite FeS2 microspheres with uniform morphology and an average size of approximately 1.1 μm were successfully synthesised via a facile and efficient microwave-assisted method. The pure phase and highly crystalline structure of the FeS2 microspheres were well confirmed via XRD, HRTEM, Raman spectroscopy and XPS. Optical absorption spectra indicate that the as-obtained pyrite FeS2 is a promising candidate material for solar absorption application. Surfactants can influence the phase, morphology and absorption property of the products. A relatively long reaction time was ideal to the formation of uniform microspheres with smooth surfaces. The microwave-assisted method can rapidly synthesise uniform pyrite FeS2 with better absorption property than the high-pressure solvothermal heating method.
Acknowledgements
This work was financially supported by the Natural Science Foundation of China (No. 51171208 and 51271201) and the Science Foundation of China University of Petroleum, Beijing (No. LLYJ-2011-41).
References
- P. P. Altermatt, T. Kiesewetter, K. Ellmer and H. Tributsch, Sol. Energy Mater. Sol. Cells, 2002, 71, 181–185 CrossRef CAS
 .
. - A. Ennaoui, S. Fiechter, H. Goslowsky and H. Tributsch, J. Electrochem. Soc., 1985, 132, 1579–1582 CrossRef CAS PubMed .
- M. Cabán-Acevedo, M. S. Faber, Y. Tan, R. J. Hamers and S. Jin, Nano Lett., 2012, 12, 1977–1982 CrossRef PubMed .
- C. Wadia, Y. Wu, S. Gul, S. K. Volkman, J. Guo and A. P. Alivisatos, Chem. Mater., 2009, 21, 2568 CrossRef CAS .
- J. Xia, J. Jiao, B. Dai, W. Qiu, S. He, W. Qiu, P. Shen and L. Chen, RSC Adv., 2013, 3, 6132–6140 RSC .
- D. Zhang, G. Wu, J. Xiang, J. Jin, Y. Cai and G. Li, Mater. Sci. Eng., B, 2013, 178, 483–488 CrossRef CAS PubMed .
- R. Morrish, R. Silverstein and C. A. Wolden, J. Am. Chem. Soc., 2012, 134, 17854–17857 CrossRef CAS PubMed .
- K. Ramasamy, M. A. Malik, M. Helliwell, F. Tuna and P. O'Brien, Inorg. Chem., 2010, 49, 8495–8503 CrossRef CAS PubMed .
- B. B. Yu, X. Zhang, Y. Jiang, J. Liu, L. Gu, J. S. Hu and L. J. Wan, J. Am. Chem. Soc., 2015, 137, 2211–2214 CrossRef CAS PubMed .
- H. Ge, L. Hai, R. R. Prabhakar, L. Y. Ming and T. Sritharan, RSC Adv., 2014, 4, 16489–16496 RSC .
- D. Wang, Q. Wang and T. Wang, CrystEngComm, 2010, 12, 3797–3805 RSC .
- Y. Bai, J. Yeom, M. Yang, S. Cha, K. Sun and N. A. Kotov, J. Phys. Chem. C, 2013, 117, 2567–2573 CAS .
- W. L. Liu, X. H. Rui, H. T. Tan, C. Xu, Q. Y. Yan and H. H. Hng, RSC Adv., 2014, 4, 48770–48776 RSC .
- X. Shi, A. Tian, X. Xue, H. Yang and Q. Xu, Mater. Lett., 2015, 141, 104–106 CrossRef CAS PubMed .
- X. H. Liao, J. J. Zhu and H. Y. Chen, Mater. Sci. Eng., B, 2001, 85, 85–89 CrossRef .
- Y. Zhao, J. J. Zhu, J. M. Hong, N. Bian and H. Y. Chen, Eur. J. Inorg. Chem., 2004, 2004, 4072–4080 CrossRef PubMed .
- S. J. Yaghmour and W. E. Mahmoud, Mater. Lett., 2013, 109, 55–57 CrossRef CAS PubMed .
- E. C. Linganiso, S. D. Mhlanga, N. J. Coville and B. W. Mwakikunga, J. Alloys Compd., 2013, 552, 345–350 CrossRef CAS PubMed .
- Y. Wang, X. Ai, D. Miller, P. Rice, T. Topuria, L. Krupp, A. Kellock and Q. Song, CrystEngComm, 2012, 14, 7560–7562 RSC .
- Y. Jung, Y. H. Son and J. K. Lee, RSC Adv., 2012, 2, 5877–5884 RSC .
- E. J. Kim and B. Batchelor, Mater. Res. Bull., 2009, 44, 1553–1558 CrossRef CAS PubMed .
- F. Long, J. He, M. Zhang, X. Wu, S. Mo, Z. Zou and Y. Zhou, J. Mater. Sci., 2015, 50, 1848–1854 CrossRef CAS .
- M. L. Li, Q. Z. Yao, G. T. Zhou, X. F. Qu, C. F. Mu and S. Q. Fu, CrystEngComm, 2011, 13, 5936–5942 RSC .
- N. Berry, M. Cheng, C. L. Perkins, M. Limpinsel, J. C. Hemminger and M. Law, Adv. Energy Mater., 2012, 2, 1124–1135 CrossRef CAS PubMed .
- L. Li, M. Cabán-Acevedo, S. N. Girard and S. Jin, Nanoscale, 2014, 6, 2112–2118 RSC .
- X. Qiu, M. Liu, T. Hayashi, M. Miyauchi and K. Hashimoto, Chem. Commun., 2013, 49, 1232–1234 RSC .
- C. Wadia, Y. Wu, S. Gul, S. K. Volkman, J. Guo and A. P. Alivisatos, Chem. Mater., 2009, 21, 2568–2570 CrossRef CAS .
- A. Ennaoui, S. Fiechter, C. Pettenkofer, N. Alonso-Vante, K. Büker, M. Bronold, Ch. Höpfner and H. Tributsch, Sol. Energy Mater. Sol. Cells, 1993, 29, 289–370 CrossRef CAS .
- S. M. Wan, F. Guo, L. Shi, Y. Y. Peng, X. Z. Liu, Y. G. Zhang and Y. T. Qian, J. Mater. Chem., 2004, 14, 2489–2491 RSC .
- Z. W. Quan, C. X. Li, X. M. Zhang, J. Yang, P. P. Yang, C. M. Zhang and J. Lin, Cryst. Growth Des., 2008, 8, 2384–2392 CAS .
- J. B. Murowchick and H. L. Barnes, Geochim. Cosmochim. Acta, 1986, 50, 2615–2629 CrossRef CAS .
- L. G. Benning, R. T. Wilkin and H. L. Barnes, Chem. Geol., 2000, 167, 25–51 CrossRef CAS .
- Y. Bi, Y. Yuan, C. L. Exstrom, S. A. Darveau and J. Huang, Nano Lett., 2011, 11, 4953–4957 CrossRef CAS PubMed .
- M. Niederberger, G. Garnweitner, F. Krumeich, R. Nesper, H. Cölfen and M. Antonietti, Chem. Mater., 2004, 16, 1202–1208 CrossRef CAS .
- E. K. Watson and W. A. Rickelton, Solvent Extr. Ion Exch., 1992, 10, 879–889 CrossRef CAS PubMed .
- J. N. Gao, Q. S. Li, H. B. Zhao, L. S. Li, C. L. Liu, Q. H. Gong and L. M. Qi, Chem. Mater., 2008, 20, 6263–6269 CrossRef CAS .
- X. H. Li, J. X. Li, G. D. Li, D. P. Liu and J. S. Chen, Chem.–Eur. J., 2007, 13, 8754–8761 CrossRef CAS PubMed .
- Z. G. Cheng, S. Z. Wang, Q. Wang and B. Y. Geng, CrystEngComm, 2010, 12, 144–149 RSC .
- Y. W. Jun, J. S. Choi and J. Cheon, Angew. Chem., Int. Ed., 2006, 45, 3414 CrossRef CAS PubMed .
- Y. D. Yin and A. P. Alivisatos, Nature, 2005, 437, 664 CrossRef CAS PubMed .
- S. M. Lee, S. N. Cho and J. Cheon, Adv. Mater., 2003, 15, 441 CrossRef CAS PubMed .
- S. Kumar and T. Nann, Small, 2006, 2, 316 CrossRef CAS PubMed .
- D. W. Wang, Q. H. Wang and T. M. Wang, CrystEngComm, 2010, 12, 755–761 RSC .
- J. Jiao, L. Chen, D. Kuang, W. Gao, H. Feng and J. Xia, RSC Adv., 2011, 1, 255–261 RSC .
- L. Zhu, B. J. Richardson and Q. Yu, Nanoscale, 2014, 6, 1029–1037 RSC .
- A. Kirkeminde, B. A. Ruzicka, R. Wang, S. Puna, H. Zhao and S. Ren, ACS Appl. Mater. Interfaces, 2012, 4, 1174–1177 CAS .
- B. Yuan, W. Luan and S. T. Tu, Dalton Trans., 2012, 772–776 RSC .
- D. Wan, Y. Wang, B. Wang, C. Ma, H. Sun and L. Wei, J. Cryst. Growth, 2003, 253, 230–238 CrossRef CAS .
- F. Sansoz, Nano Lett., 2011, 11, 5378–5382 CrossRef CAS PubMed .
- G. C. Xi, C. Wang and X. Wang, Eur. J. Inorg. Chem., 2008, 2008, 425–431 CrossRef PubMed .
- Y. D. Li, H. Wang, X. F. Wang and J. B. Bai, J. Cryst. Growth, 2009, 311, 3445–3450 CrossRef PubMed .
- X. Wu, K. W. Li and H. Wang, J. Alloys Compd., 2009, 487, 537–544 CrossRef CAS PubMed .
- M. Wang, C. Xing, K. Cao, L. Zhang, J. Liu and L. Meng, J. Mater. Chem. A, 2014, 2, 9496–9505 CAS .
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 











