
Single and double intramolecular proton transfers in the electronically excited state of flavone derivatives†
I. E. Serdiuk*ab and
A. D. Roshalb
aDepartment of Chemistry, University of Gdańsk, Gdańsk, 80-308 Poland. E-mail: illia.serdiuk@gmail.com
bInstitute of Chemistry, V. N. Karazin Kharkiv National University, Kharkiv, 61022 Ukraine
First published on 23rd November 2015
Abstract
In an attempt to create a flavone derivative able to take part in Excited State Intramolecular Double Proton Transfer (ESIDPT), we synthesized two carbonyl derivatives of 3,7-dihydroxyflavone, both containing two different proton-transfer sites as well as related carbonyl derivatives of 3-hydroxyflavone and 7-hydroxyflavone. All the examined hydroxyflavones were found to participate in the Excited State Intramolecular Proton Transfer (ESIPT). ESIPT which involves 3-hydroxyl and 4-carbonyl groups was found to have a higher barrier compared to ESIPT involving 7-hydroxyl and 6/8-carbonyl fragments. According to the data presented, 3,7-dihydroxy-2-phenyl-6-(3-phenylpropanoyl)-4H-chromen-4-one undergoes a two-stage ESIDPT with formation of an intermediate tautomer. This kind of ESIDPT leads to a tautomeric form with an abnormally low rate of radiative deactivation of the excited state, which conditions low fluorescence quantum yield. The behavior of 3,7-dihydroxy-4-oxo-2-phenyl-4H-chromene-8-carbaldehyde in the electronically excited state is similar to 3-hydroxyflavone derivatives, thus we conclude the occurrence of a single ESIPT in this compound.
Introduction
Excited State Intramolecular Proton Transfer (ESIPT) represents one of the fundamental reactions in photochemistry. The transformation occurs most often in compounds which contain fragments participating in keto–enol and imine–amine tautomerization reactions connected by the intramolecular hydrogen bond (IHB).1–3 Due to its very low energetic barrier ESIPT is known to be ultrafast at various conditions, which allows it to successfully compete with other deactivation pathways of the electronically excited state, such as radiative deactivation. Followed by a reverse proton transfer in ground state, ESIPT conditions high photostability of the compounds, and specific fluorescent properties namely large red-shift of emission, which is practically insensitive to reabsorption effect and very sensitive to external hydrogen bonding. ESIPT fluorophores have various applications in optoelectronic materials2 and as biological markers and probes.3Excited State Intramolecular Double Proton Transfer (ESIDPT) is a very rare process, which involves two proton-transfer (PT) sites in one molecule. Hypothetically, ESIDPT can be as useful as ESIPT or even more, if one expects higher energy losses of the electronically excited molecule before it emits light. Various bifunctional organic compounds were designed and synthesized, including derivatives of salicylic acid,4 oxazoles,5–7 and chromones,8–11 and subsequently investigated from the point of view of ESIDPT by steady-state and time-dependent absorption and fluorescent spectroscopies, as well as ab initio and semiempirical computational techniques. Generally, it was found that after excitation these compounds undergo single ESIPT, while occurrence of ESIDPT is much less favorable. It should be noticed that so far scientists' attention was mostly paid to compounds with axial symmetry and the same two PT sites.
In this article we present synthetic routine and results of spectroscopy investigations of two novel asymmetric flavone derivatives: 3,7-dihydroxy-2-phenyl-6-(3-phenylpropanoyl)-4H-chromen-4-one (1a) and 3,7-dihydroxy-4-oxo-2-phenyl-4H-chromene-8-carbaldehyde (2a) (Chart 1). Both of the investigated compounds contain two different PT sites able to participate in keto–enol tautomerization reactions. The first site is represented by a hydroxyl group in position 7 and a carbonyl fragment in ortho position: 3-phenylpropanoyl in position 6 (1a) and formyl in position 8 (2a). The second site includes hydroxyl and carbonyl group in positions 3 and 4, respectively. These fragments are similar to PT sites of ortho-hydroxybenzaldehyde and flavonol (3-hydroxy-2-phenyl-4H-chromen-4-one) derivatives, both known to participate in ESIPT.12 Compounds 1a and 2a can possibly exist in 4 tautomeric forms (Charts 2 and 3), transforming one to another via intramolecular proton transfer reactions. Forms T6 (1a) and T8 (2a) can be produced via PT involving hydroxyl groups in position 7, T4 can appear in the result of PT involving hydroxyl groups in position 3, and DT can be formed in the result of double PT. The related compounds 7-hydroxy-2-phenyl-6-(3-phenylpropanoyl)-4H-chromen-4-one (1b), 3-hydroxy-7-methoxy-2-phenyl-6-(3-phenylpropanoyl)-4H-chromen-4-one (1c), and 7-hydroxy-4-oxo-2-phenyl-4H-chromene-8-carbaldehyde (2b), 7-hydroxy-3-methoxy-4-oxo-2-phenyl-4H-chromene-8-carbaldehyde (2c), which contain only one hydroxyl group were used as models for investigation of single PT reactions as well as properties of T6, T4, and T8, respectively. Compounds 7-methoxy-2-phenyl-6-(3-phenylpropanoyl)-4H-chromen-4-one (1d) and 7-methoxy-4-oxo-2-phenyl-4H-chromene-8-carbaldehyde (2d), which do not contain hydroxyl groups, were used for investigations of the target compounds' behavior in the absence of PT reactions. Taking into account that 1a–c and 2a–c can possibly exist in anionic forms besides neutral ones, spectral behavior of these compounds was investigated in the presence of DBU. This helped to define origin of absorbance and fluorescence of 1a and 2a.
 | ||
| Chart 1 Canonical structures of 1a–d and 2a–d with numbering of atoms indicated. | ||
 | ||
| Chart 2 Structures of possible tautomeric forms (N, T4, T6, and DT) of 1a (Chart 1). | ||
 | ||
| Chart 3 Structures of possible tautomeric forms (N, T4, T8, and DT) of 2a (Chart 1). | ||
Results and discussion
Synthesis
In an attempt to obtain a derivative of 3,7-dihydroxyflavone with a carbonyl fragment in position 6 we firstly made an attempt to synthesize 6-acetyl-3,7-dihydroxy-2-phenyl-4H-chromen-4-one starting from the commercially available 1,1′-(4,6-dihydroxy-1,3-phenylene)bisethanone. However, reaction of the latter with equimolar quantity of benzaldehyde and other aldehydes in alkalinic media led to a mixture of mono- and dichalcone derivatives,13 which were difficult to separate. In order to eliminate formation of the undesired dichalcone product, 1,1′-(4,6-dihydroxy-1,3-phenylene)bisethanone derivatives were designed with substituent(s) in one of the α positions (5, Scheme 1). Most probably, 1-(5-acetyl-2,4-dihydroxyphenyl)-2,2-dimethylpropan-1-one (5b) would react with only one equivalent of benzaldehyde and give a monochalcone derivative. Preparation of 5b was, however, problematic: the pivaloyl fragment of 1-(2,4-dihydroxyphenyl)-2,2-dimethylpropan-1-one (4b), prepared by reaction of resorcinol with pivaloyl chloride in hot boron trifluoride etherate was unstable in the presence of Lewis acids14 utilized in further acylation step and only 1-(3-acetyl-2,4-dihydroxyphenyl)-2,2-dimethylpropan-1-one (6b) was isolated in a low yield. 1-(2,4-Dihydroxyphenyl)-3-phenylpropan-1-one (4a, Scheme 1) on the other hand, obtained in the same conditions from resorcinol and 3-phenylpropionic acid, gave the desired 1-(5-acetyl-2,4-dihydroxyphenyl)-3-phenylpropan-1-one (5a) in the reaction with acetic anhydride in the presence of ZnCl2 in a good yield (51%) and minor impurity of 1-(3-acetyl-2,4-dihydroxyphenyl)-3-phenylpropan-1-one (6a) (8%), which was separated by column chromatography. 5a reacted with equimolar quantity of benzaldehyde in aqueous N-methyl-pyrrolidone (NMP) in the presence of KOH leading to the chalcone derivative 1e in 48% yield. When 1e was treated with H2O2 in MeOH in the presence of NaOH (Algar–Flynn–Oyamada reaction conditions) the desired 1a was not identified in the reaction mixture. This phenomenon can be explained by existence of 1e at such conditions in doubly deprotonated form, as in the case of (2E)-1-(2,4-dihydroxyphenyl)-3-phenylprop-2-en-1-one,15 which does not transform to the corresponding 3-hydroxyflavone derivative. 1e was then converted to the flavone derivative 1b in hot DMSO in the presence of I2 (yield 53%), which after protection of the hydroxyl group by dimethylsulfate was oxidized with dimethyldioxirane, generated in situ by Oxone© and acetone in water–chloroform (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) suspension at 0–5 °C. The obtained epoxide of 1d was then hydrolyzed by TFA without isolation, what produced 3-hydroxyflavone derivative 1c in 91% yield. Finally, 1a was obtained by reaction of 1c with boron tribromide in CH2Cl2 (yield 87%). The applied synthetic route allowed for obtaining not only the target compound 1a, but also the relative ones 1b, 1c and 1d, which were used as models in the further investigations.
:1, v/v) suspension at 0–5 °C. The obtained epoxide of 1d was then hydrolyzed by TFA without isolation, what produced 3-hydroxyflavone derivative 1c in 91% yield. Finally, 1a was obtained by reaction of 1c with boron tribromide in CH2Cl2 (yield 87%). The applied synthetic route allowed for obtaining not only the target compound 1a, but also the relative ones 1b, 1c and 1d, which were used as models in the further investigations.
 | ||
| Scheme 1 Synthesis of 1a–d. | ||
For synthesis of 2a (Scheme 2), 3-hydroxy-7-benzyloxyflavone (10) was chosen as a precursor, synthesized from commercially available 1-(2,4-dihydroxyphenyl)ethanone (7), according to the procedure described earlier:15 7 was treated with benzyl chloride, leading to 8a, then converted to chalcone derivative 9a with benzaldehyde in basic NMP, which was then oxidatively cyclized in basic MeOH in the presence of H2O2 (Algar–Flynn–Oyamada reaction). 10 was subsequently alkylated with dimethylsulfate, followed by cleavage of benzyl group in HBr solution in acetic acid to yield 12. Treatment of 12 with excess of hexamethylenetetramine in boiling glacial acetic acid (Duff reaction)16 gave 2c in 59% yield. Finally, 2a was obtained by reaction of 2c with boron tribromide. Relative compound 2b was prepared from 7-hydroxyflavone (14) in the Duff reaction conditions in 67% yield. 14 was prepared starting from 7, using methyl protection for hydroxyl group via stages described in ref. 17. 2d was obtained from 2b by methylation with dimethylsulfate in CH3CN.
 | ||
| Scheme 2 Synthesis of 2a–d. | ||
Steady state electronic absorption spectroscopy
The compounds investigated exhibit absorption in methylcyclohexane in the 250–400 nm region (Table 1, Fig. 1a and b), which supposedly corresponds to N form (Charts 2 and 3), usually the most stable in the ground state of flavones. Spectral properties of anionic forms of the compounds were investigated in DBU solutions. Positions of the long-wavelength bands of neutral and charged species depend on the nature and position of substituents.| Compd | cDBU (M) | T (K) | λabs (nm) | λex (nm) | λfl (nm) | ΔνSt (cm−1) | Compd | cDBU (M) | T (K) | λabs (nm) | λex (nm) | λfl (nm) | ΔνSt (cm−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a cDBU – molar concentration of DBU; λabs – position of the maximum of the long-wavelength band in absorption spectra; λex – position of the maximum of the long-wavelength band in fluorescence excitation spectra; λfl – position of the maximum in the fluorescence emission spectra (for bands with vibrational modes λfl represents center-weighted position of a band); ΔνSt – Stokes shifts, calculated as a difference between (λex)−1 and (λfl)−1.b Phosphorescence maxima.c Maxima of the long-wavelength bands obtained by separation. | |||||||||||||
| 1a | 298 | 352 | 348 | 562 | 10940 |
2a | 298 | 339 | 337 | 551 | 11530 |
||
| 77 | 378 | 563 | 8690 | 77 | 335 | 415 | 5750 | ||||||
| 338 | 535 | 10890 |
|||||||||||
| 1 × 10−4 | 298 | 430 | — | — | — | 1 × 10−4 | 298 | 375c | — | — | — | ||
| 77 | 446 | 543 | 4000 | 77 | — | — | — | ||||||
| 1 × 10−3 | 298 | 392 | — | — | — | 1 × 10−3 | 298 | 402 | 402 | 565 | 7180 | ||
| 77 | 446 | 543 | 4000 | 77 | 404 | 541 | 6270 | ||||||
| 1b | 298 | 324 | 325 | 546 | 12450 |
2b | 298 | 336 | 338 | 505 | 9780 | ||
| 77 | 325 | 530 | 11900 |
77 | 337 | 484 | 9010 | ||||||
| 1 × 10−4 | 298 | 412 | 405 | 526 | 5680 | 1 × 10−4 | 298 | 392 | 390 | 518 | 6320 | ||
| 77 | 404 | 489 | 4300 | 77 | 392 | 485 | 4900 | ||||||
| 1c | 298 | 334 | 336 | 542 | 11320 |
2c | 298 | 331c | 332c | 513 | 10630 |
||
| 77 | 337 | 424 | 6090 | 77 | 332c | 481 | 9330 | ||||||
| 338 | 540 | 11070 |
|||||||||||
| 1 × 10−4 | 298 | 413 | 415 | 502 | 4180 | 1 × 10−4 | 298 | 381 | 381 | 505 | 6480 | ||
| 77 | — | — | — | 77 | 382 | 475 | 5130 | ||||||
| 1d | 298 | 295 | — | — | — | 2d | 298 | 306 | — | — | — | ||
| 77 | 496b | 77 | 538b | ||||||||||
 | ||
| Fig. 1 Steady-state absorption spectra at 298 K: 1a–d (a) and 2a–d (b) in methylcyclohexane; 1a–c (c) and 2a–c (d) in 10−4 M DBU solution in methylcyclohexane. | ||
The long-wavelength absorption bands of 1d and 2d are positioned at 295 and 306 nm, respectively. The bands of 1b and 2b, which contain IHB, are shifted batochromically to 324 and 336 nm, respectively (Table 1, Fig. 1a and b). Compared to 7-hydroxyflavone, whose absorption band at the same conditions is positioned at 291 nm,18 1b and 2b exhibit batochromic shift, caused by polarization of the hydroxyl group at position 7 due to IHB. Compared to 1b, the long-wavelength absorption band of 1c is batochromically shifted to 334 nm (Table 1, Fig. 1a), which is caused by influence of the hydroxyl group in position 3. Similar spectral effect was observed previously in the case of 3-hydroxyflavone derivatives.19 2c exhibits complex absorption spectra (Fig. 1b), its long-wavelength band appears in the 325–350 nm range, similarly to 2b. Methoxy group at position 3 thus has minimal effect on the S0–S1 transition energy, which is most probably due to high value of the torsion angle between chromone moiety and methoxy group, as was observed in the case of 3-methoxyflavone derivatives.20
1a (352 nm) and 2a (339 nm) exhibit the most batochromically shifted absorption among the compounds investigated (Table 1, Fig. 1a and b). 3,7-Dihydroxyflavone in the same conditions exhibits absorption at 337 nm (unpublished data), close to that of 2a. Thus the carbonyl substituent in position 6 affects the S0–S1 transition energy of 3,7-dihydroxyflavone much more considerably than in position 8.
The long-wavelength absorption bands of the anionic species of the hydroxyflavones investigated, which appear in DBU solutions, are considerably shifted batochromically relative to the neutral ones. Monoanionic species A7 (Chart 4), formed by deprotonation of the hydroxyl group at position 7 of 1b and 2b, absorb light at 412 and 392 nm, respectively. Compared to anion of 7-hydroxyflavone18 this corresponds to the batochromic shift values of 3240 and 2000 cm−1, respectively, due to electron-withdrawing effect of the carbonyl substituents. More than 1.5 lower value of the shift in the case of 2b compared to 1b indicates less effective stabilization of A7 by the carbonyl substituent in position 8 compared to position 6. Absorption maximum of the similar species of 2c is shifted hypsochromically to 381 nm relative to 2b, which indicates that due to electron-releasing effect methoxy group at position 3 increases the S0–S1 transition energy in A7. The long-wavelength absorption band of the monoanionic species A3, formed by deprotonation of the hydroxyl group at position 3 of 1c, is centered at 413 nm (Chart 4).
 | ||
| Chart 4 Possible monoanionic forms of 1a–c and 2a–c. | ||
A monoanionic form of 1a, which appears in 10−4 M DBU solution, absorbs light at 430 nm (Table 1, Fig. 1c). Deprotonation of the second hydroxyl group leads to a 2680 cm−1 hypsochromic shift. On the other hand, monoanionic species of 2a absorb light at 375 nm, while further deprotonation leads to a 1790 cm−1 batochromic shift (Table 1, Fig. 1d). The described differences in the spectral behavior of anions of 1a and 2a may indicate different order of deprotonation of hydroxyl groups and formation of different monoanionic forms (Chart 4).
Steady state fluorescence emission and excitation spectroscopy
Since 1a and 2a have two different PT sites which can participate in ESIPT simultaneously, the experimental investigations of their electronically excited state behavior are complicated. In order to understand the features of each PT site, we investigated the related compounds. 1d and 2d enabled investigation of the N* forms properties in the absence of IHB and ESIPT. 1b and 2b, c provided information concerning behavior of hydroxyl group in position 7 with ortho-carbonyl substituents as well as spectral features of T6* and T8* forms, respectively. 1c enabled investigation of ESIPT involving hydroxyl and carbonyl group in positions 3 and 4, respectively. Fluorescent behavior of 1b, c and 2b, c in DBU solutions provided information concerning features of monoanionic forms A7* and A3* as well as deprotonation order of hydroxyl groups in 1a and 2a. The conclusions on the excited state behavior of 1a and 2a were thus made based on a comparison of their spectral features with the related compounds.At 298 K all the hydroxyflavones investigated are characterized by a single-band fluorescence spectra with the abnormally large Stokes shifts (Table 1, Fig. 2 and S2†) indicating occurrence of efficient ESIPT. 1b and 2b, c exhibit broad emission bands centered at 546, and 505, 513 nm, respectively (Table 1, Fig. 2c, d and f) with fluorescence quantum yields less than 1%. In contrast to 1b, vibronic structure of the 2b and 2c bands is visible. When the 1b and 2b, c solutions are cooled to 77 K, their fluorescence intensity increases, maxima shift to 530 and 484, 481 nm, respectively, and bands become narrower (Table 1, Fig. 2c, d and f).
 | ||
| Fig. 2 Steady-state fluorescence spectra of 1a (a), 2a (b), 1b (c), 2b (d), 1c (e) and 2c (f) in methylcyclohexane at 298 K and 77 K. | ||
Fluorescence of anionic forms A7* appear in DBU solutions of 1b and 2b, c with maxima near 526 and 518, 505 nm, respectively. At 77 K their intensity increases and maxima shift to 489 and 485, 475 nm, respectively. In contrast to neutral solutions, vibronic structure is not observed in the case of the 2b, c fluorescence in DBU solutions.
As can be concluded from the 3D steady-state fluorescence spectra and similarity of the long-wavelength bands in absorption and fluorescence excitation spectra at 298 K and 77 K (Table 1, Fig. S2c, d, f and S3c, d, f†), the described emission of 1b and 2b, c in neutral solutions originates from N species in the ground state. Generally, shape of the emission bands as well as spectral changes under cooling of neutral solutions differ from those of DBU ones, which evidences that fluorescence of anionic forms is absent at such conditions. Most probably, the T6* and T8* species (Charts 2 and 3), respectively, produced via ESIPT in N* after excitation (Scheme 3), are responsible for fluorescence of these compounds in methylcyclohexane.
 | ||
| Scheme 3 ESIPT in 1b and 2b, c. | ||
Neither 1b nor 2b, c exhibit phosphorescence at 77 K. As was discussed above, the long-wavelength absorption bands of 1b and 2b, c are considerably red-shifted compared to 1d and 2d due to IHB. This can emphasize decrease of the π–π* state energy, which is more sensitive to changes in electronic structure of chromofores compared to the n–π* one. Increase of the gap between π–π* and n–π* states prevents intersystem crossing and appearance of phosphorescence and should moreover enhance fluorescence of the N* species, as in the case of 4′-methoxyflavone.21 Absence of the N* fluorescence neither at 298 K nor 77 K evidences very fast ESIPT in 1b and 2b, c.
Very low intensive fluorescence of anion A3* appear at 502 nm in DBU solutions of 1c at 298 K (Table 1, Fig. 2e). At 77 K no fluorescence of 1c in DBU solutions was detected.
Most probably, the T4* species, produced via ESIPT in N* after excitation (Scheme 4), are responsible for the long-wavelength fluorescence emission in neutral solutions of 1c both at 298 and 77 K. The observed blue-shifted emission at 77 K most probably originates from N* produced by direct excitation. Appearance of the N* fluorescence under cooling indicates existence of energetic barrier for the ESIPT reaction N* → T4*, previously observed for 3-hydroxyflavone and relative compounds.22,23 The substituents' effect can cause stabilization of the N* form in the case of 1c compared to 3-hydroxyflavone, what can explain increase of the N* → T4* reaction barrier. The A3* fluorescence, which was assumed to appear in solutions of 3-hydroxyflavone in hydrocarbons at 77 K due to the presence of impurities of protic substances,24 can not have noticeable impact in fluorescence of 1c due to very low emission intensity.
 | ||
| Scheme 4 ESIPT in 1c. | ||
| Compd | Form | τ (ns)/f (%) | kf × 10−7 (s−1) | kd × 10−9 (s−1) | φ (%) |
|---|---|---|---|---|---|
| a τ – lifetime of the electronically excited form; f – fractional contribution, calculated as fi = Aiτi/∑Aiτi, where Ai are the pre-exponential factors; kf – rate constant of radiative deactivation; kd – rate constant of non-radiative deactivation; φ – quantum yield of fluorescence. | |||||
| 1a | DT* | 5.89 ± 0.02/94 | 0.02 | 0.17 | 0.11 |
| T6* | 0.025 ± 0.005/6 | ||||
| 1b | T6* | 0.473 ± 0.007 | 2.11 | 2.09 | 1.0 |
| 1c | T4* | 2.19 ± 0.03 | 5.30 | 0.40 | 11.6 |
| 2a | T4* | 1.98 ± 0.04 | 0.81 | 0.50 | 1.6 |
| 2b | T8* | 0.377 ± 0.008 | 0.58 | 2.65 | 0.22 |
| 2c | T8* | 0.415 ± 0.004 | 0.65 | 2.40 | 0.27 |
1a demonstrates intensive fluorescence at 543 nm in DBU solutions under cooling to 77 K (Fig. 2a). The excitation spectrum has maxima at 446 nm, which resembles absorption spectrum of monoanionic form, observed at 298 K (Table 1, Fig. S3a†). The fluorescence behavior of the 1a monoanionic species at 77 K is similar to 1b, but not 1c, which can indicate that A7* species are responsible for the 1a fluorescence at 77 K in the presence of DBU. The excitation spectrum of 1a in neutral solution at 77 K matches absorption/excitation of neither its A7* species nor A3* ones of 1c. Dianionic species, which are present in the 10−3 M DBU solution, are either non-fluorescent or have low fluorescence intensity relative to A7* species at 298 and 77 K. Formation of the doubly charged dianion in neutral methylcyclohexane should be extremely unfavorable. Based on these data we conclude that deprotonation is not the reason of the batochromic shift in the excitation spectrum of 1a at 77 K.
Similarly to 3,7-dihydroxyflavone,19,25 in the ground state of 1a hydroxyl groups and carbonyl group at position 4 should be of low acidity and basicity, respectively, thus formation of tautomeric species T4 is unfavorable. As follows from acid–base properties of acetophenones,26 basicity of the carbonyl group at position 6 of 1a should be also very low, thus formation of T6 as well as DT in the ground state is unfavorable too. Taking into account that concentrations of the investigated solutions of 1a were below 5 × 10−5 M, formation of aggregates is of low probability. These considerations support the mentioned above conclusion on origination of 1a fluorescence from N species in the ground state. Hypothetically, the mentioned above batochromic shift of the excitation spectra of 1a under cooling can be explained by strengthening of both of its hydrogen bonds, which can decrease relative energy of the excited Franck–Condon state and thus decrease the S0–S1 transition energy of N species.
Based on the data presented, one can notice the basic difference in the spectral behavior of 1a and 1c such as much lower fluorescence quantum yield (Table 2), absence of the N* emission at 77 K in the case of 1a and different behavior of monoanionic forms (Fig. 2a and e). This can indicate that 1a does not exist in T4* form. Compared to 1b, 1a has a tenfold lower fluorescence quantum yield, however, behavior of both compounds at 77 K is generally similar in both neutral and DBU methylcyclohexane solutions (Fig. 2a and c). These observations can indicate that hydroxyl group at position 7 has prior influence on the 1a properties due to its higher acidity compared to hydroxyl group at position 3. The fluorescence of 1a in the neutral solutions can be, thus, generated by either T6*, DT* or both, but not the T4* form (Scheme 5).
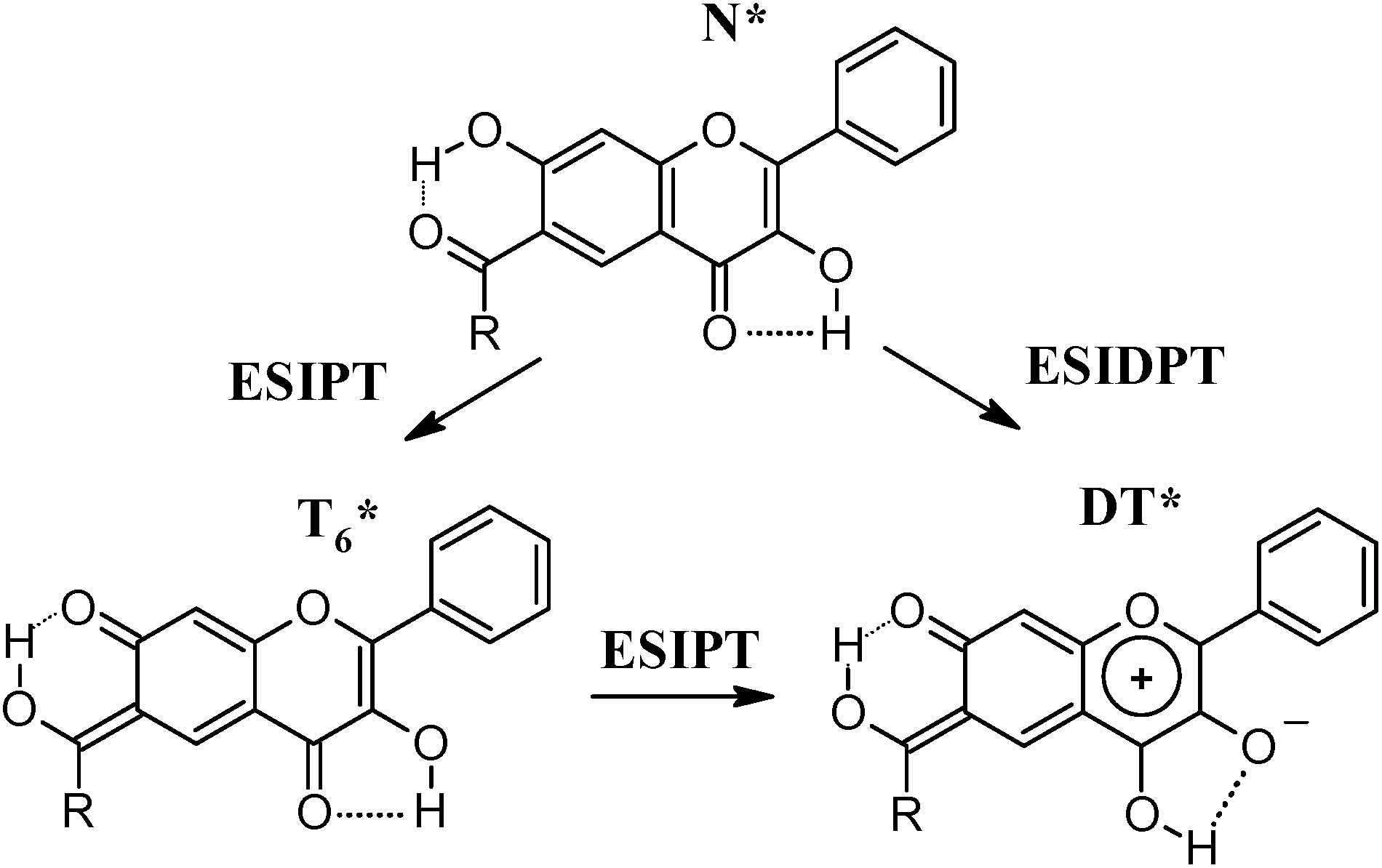 | ||
| Scheme 5 ESIPT and ESIDPT in 1a. | ||
Monoanionic forms of 2a, which according to the described above absorption spectra appear in the 10−4 M DBU solution, are fluorescent neither at 298 K nor 77 K. This phenomenon, in contrast to intensive fluorescence of the A7* forms of 2b and 2c, can indicate that hydroxyl group of position 3 of 2a is deprotonated first, producing A3* form. Different from 1a deprotonation order of hydroxyl groups in 2a can indicate that acidity of the hydroxyl group at position 7 is for less extent affected by the carbonyl substituent at position 8 than at position 6. This conclusion correlates with the discussed above influence of the substituents on the A7 spectral properties of 1b and 2b.
Broad fluorescence band appear in spectrum of 2a in the 10−3 M DBU solution at 565 nm at 298 K and shifts to 541 nm at 77 K. According to the excitation spectrum the emission originates from dianionic form.
Compared to 2b and 2c, in neutral solutions 2a has a few times higher fluorescence quantum yield and exhibits N* fluorescence emission at 77 K, which indicates absence of T8* form in the excited state of 2a. Even though a model compound for T4* form of 2a was not available in this study, spectral properties including positions of the fluorescence emission and excitation maxima together with behavior at 77 K of 2a and 1c are very similar. The fluorescence of 2a can be, thus, generated by either T4* (Scheme 6), DT* forms or both, but not the T8*, as hydroxyl group at position 3 has prior influence on the 2a properties.
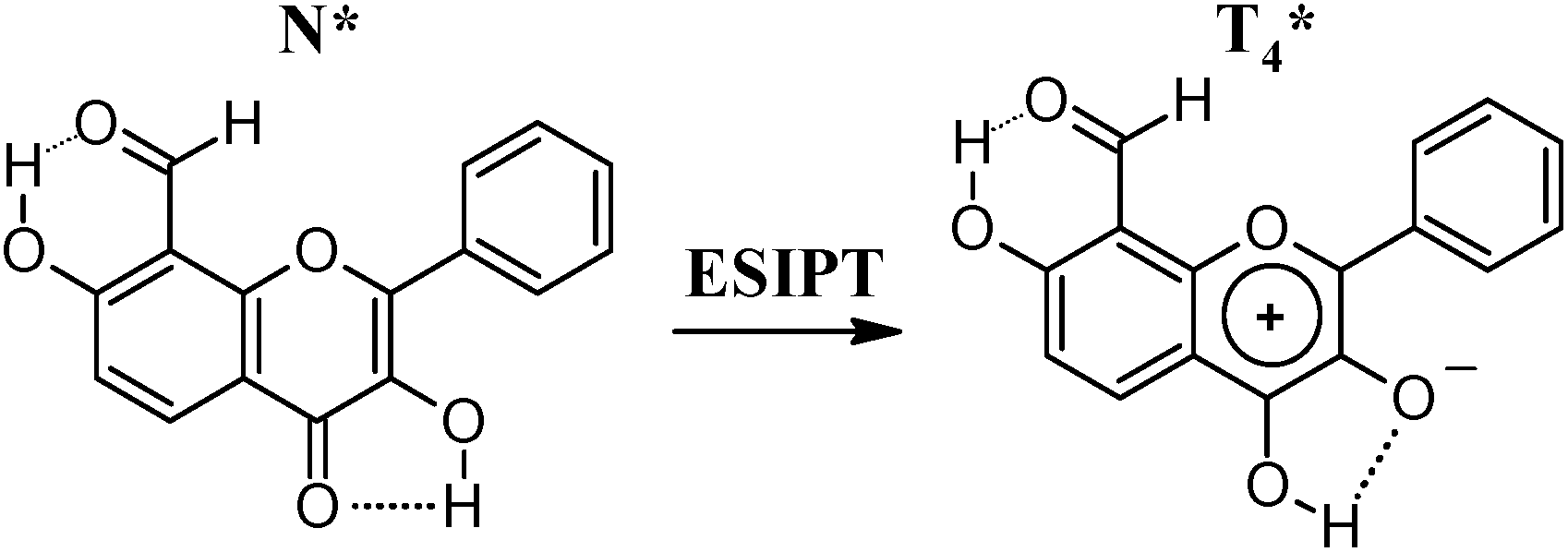 | ||
| Scheme 6 ESIPT in 2a. | ||
Time-resolved fluorescence spectroscopy
Fluorescence decays of 1b and 2b, c in hexane are biexponential without rise components in the investigated time domain. The major components have lifetimes of less than 0.5 ns (Table 2, Fig. 3a and b), while the minor ones have very low intensity and lifetimes near 1.5–3.0 ns. Precise determination of the latter components' lifetimes is complicated. The major components most probably characterize decay of T6* and T8* species, respectively. Appearance of the minor components may indicate co-existence of T6* and T8* species in two isomers, different as regards the PT sites conformations. Based on the assumption that the main deactivation pathway of the N* forms of 1b and 2b, c is ESIPT, we calculated rates of radiative (kf) and non-radiative deactivation (kd) for their T6* and T8* forms (Table 2) using the major components' lifetimes. According to the obtained values, non-radiative processes with a summary rate of 2.1–2.7 × 109 s−1 are the main deactivation pathway for both types of forms. T6* form of 1b is characterized by higher kf value compared to T8* of 2b, c, which conditions higher quantum fluorescence yield of the former (Table 2). | ||
| Fig. 3 Fluorescence decays of 1a–c (a) and 2a–c (b) in hexane at 298 K recorded at the fluorescence maxima. | ||
In hexane at 550 nm, 1c exhibits a monoexponential decay of fluorescence with 2.19 ns lifetime (Table 2, Fig. 3a) and no rise component in the investigated time domain, corresponding to deactivation of the T4* species. The kf value of T4*, calculated based on the assumption that the main deactivation pathway of the N* form is ESIPT, is the highest among the compounds investigated, what together with low kd value conditions the highest quantum yield of fluorescence (Table 2).
Fluorescence decay of 1a at 560 nm differs from the other compounds investigated: it contains two decay components with lifetimes of ∼25 ps and 5.3 ns (Table 2, Fig. 3a). Due to a drastic difference in the lifetimes, the long-living species have the overwhelming impact in 1a fluorescence quantum yield (94%), which enables estimation of their kf and kd values (Table 2). The obtained kf value is extremely low and unique among the ones of 1b and 1c, which implies that the long-living species are neither T4* nor T6*. As formation of T4* species in the excited state of 1a was suggested to be unlikely according to the steady-state fluorescence investigations, we consider that the long decay corresponds to DT* and the fast decay corresponds to T6*. The observed broadening of the 1a fluorescence emission spectra at 77 K (Fig. 2a) can thus be explained by co-existence of T6* and DT* forms. Most probably, that the T6* species are produced by ultrafast ESIPT with no or very small energetic barrier, similarly to 1b, since no N* emission is observed even at 77 K. Considering similar structure and close spectral properties of T6* forms of 1a and 1b, their rates of radiative and non-radiative deactivation should be of the same ranges. Much faster decay of the T6* species of 1a can indicate existence of an additional efficient dark deactivation pathway, which we suppose is ESIPT resulting in DT*. If one suggests that the main excited state deactivation route of T6* is ESIPT, the rate constant of the second stage should be near 4 × 1010 s−1. The ESIDPT tautomer is, thus, most probably formed via two single ESIPT reactions: N* → T6* → DT* (Scheme 5). The second stage should have an energetic barrier, as it is detectable in the sub-nanosecond time domain. Under cooling the T6* → DT* transformation should be suppressed, thus T6* should have higher impact to fluorescence of 1a.
Simultaneous ESIDPT (N* → DT*), if it occurs, should not be accompanied by formation of intermediate species (Scheme 5), which contradicts the observed experimental evidences of T6* formation at both 298 and 77 K. Simultaneous ESIDPT can hardly be a barrierless process, because the observed ESIPT involving hydroxyl group at position 3 proceed with an energetic barrier. Therefore, at low temperatures, N* fluorescence of 1a would be registered, which is not the case. We thus conclude, that possibility of DT* formation via concerted mechanism, is less likely.
2a in hexane exhibits a monoexponential decay of fluorescence with lifetime of 1.98 ns with no rise component (Fig. 3b). Compared to 2b, c, deactivation rates of 2a differ a few times (Table 2), which supports the suggestion of absence of T8* form in its excited state, based on the steady-state fluorescence investigation. Generally, spectral and kinetic properties of 2a resemble the T4* form of 1c, a difference between kf values may be caused by substitution effect. The DT* species if they appear, should be formed via ESIPT in T4*. Taking into account the observed evidence of much lower energetic barrier of ESIPT taking place in PT site including hydroxyl group in position 7, transformation T4* → DT* should be too fast to be detected by the techniques used. Formation of DT* via concerted ESIDPT seems to us unlikely due to differences of ESIPT barriers of two sites. The discussed above different deprotonation order in 1a and 2a can be the main reason of different behavior of these compounds. The following experimental and theoretical investigations may probably allow for verification of these suggestions.
Experimental
Reagents of relevant grade for syntheses and spectroscopic investigations were purchased from Sigma-Aldrich. Identity of the investigated compounds was confirmed with H1-NMR, C13-NMR and MALDI TOF MS, their purity was controlled with TLC and elemental analysis. NMR spectra were recorded on 500 MHz 1H (125 MHz 13C) or 200 MHz 1H spectrometers with trimethylsilane as reference. Mass spectra were obtained on MALDI-TOF MS Bruker Daltonics mass spectrometer. Chromatography was performed on silica gel (230–400 mesh) or Waters HPLC chromatograph, TLC was conducted on Merck 60 F254 silicagel plates in appropriate eluents. Elemental analysis was performed on an Elementar Vario El Cube CHNS analyzer.Absorption and fluorescence, phosphorescence spectra were recorded on a Perkin-Elmer Lambda UV/vis 40 spectrophotometer and Varian Cary Eclipse Fluorescence Spectrophotometer, respectively. Fluorescence and phosphorescence emission and excitation spectra were corrected on the instrumental sensitivity. Fluorescence decay curves were measured on a FluoTime 300 fluorescence lifetime spectrometer equipped with a compact emission monochromator, a TimeHarp 300E TCSPC device (minimal time resolution 4 ps), a PLS 340 LED-head for sub-nanosecond pulse driven by a PDL 820 device and a MCP-PMT photomultiplier (type R3809U-50) (PicoQuant GmbH, Germany) controlled by EasyTau system software.
Investigations of spectral properties were carried out in methylcyclohexane solutions with concentrations 1–5 × 10−5 M. Investigations of anionic forms were carried out in the methylcyclohexane solutions of DBU (1,8-diazabicyclo[5.4.0] undec-7-ene). Measurements at 77 K were held in FL-1013 liquid nitrogen dewar assembly using quartz 10 × 10 mm cuvettes with a stopper. Time-resolved fluorescence investigations were held in hexane. The solvents were treated with LiAlH4 and distilled prior use in order to eliminate trace amounts of water. The compounds were additionally dried before measurements at 373 K under reduced pressure.
Fluorescence quantum yields were determined relative to quinine sulphate solution in 1 M H2SO4.27 Rates of the excited state deactivation were calculated according to equations:28
| φ = τkf, | (1) |
where φ – quantum yield of fluorescence, τ – lifetime of the electronically excited form; kf – rate constant of radiative deactivation; kd – rate constant of non-radiative deactivation.

Synthesis
7-Hydroxy-2-phenyl-4H-chromen-4-one (14) was prepared according to ref. 17, its chemical physical properties correspond to the ones described in literature. Synthetic procedure and results of analysis for 7-(benzyloxy)-3-hydroxy-2-phenyl-4H-chromen-4-one (10) were described previously.154a: White moist solid (2.19 g, 95%), 1H NMR (500 MHz, DMSO-d6, δ): 2.91 (t, 2H, J = 7.6 Hz), 3.23 (t, 2H, J = 7.6 Hz), 6.23 (d, 1H, J = 2.2 Hz), 6.34 (dd, 1H, J = 2.2 Hz, J = 8.9 Hz), 7.17 (m, 1H, J = 4.3 Hz), 7.26 (d, 4H, J = 4.3 Hz), 7.80 (d, 1H, J = 8.9 Hz), 10.60 (broad s, 1H), 12.57 (s, 1H). Mass spectrum, m/z: 243.3 [M + H]+, 265.1 [M + Na]+, 281.0 [M + K]+.
4b: White moist solid (1.88 g, 97%), 1H NMR (200 MHz, DMSO-d6, δ): 1.30 (s, 9H), 6.25 (d, 1H, J = 2.4 Hz), 6.32 (dd, 1H, J = 2.4 Hz, J = 8.9 Hz), 7.80 (d, 1H, J = 8.9 Hz), 10.42 (s, 1H), 12.57 (s, 1H). Mass spectrum, m/z: 195.2 [M + H]+, 217.2 [M + Na]+.
5a: White moist solid (1.17 g, 51%), 1H NMR (200 MHz, DMSO-d6, δ): 2.61 (s, 3H), 2.92 (t, 2H, J = 7.6 Hz), 3.43 (t, 2H, J = 7.6 Hz), 6.36 (s, 1H), 7.18 (m, 1H, J = 4.4 Hz), 7.25 (d, 4H, J = 4.4 Hz), 8.39 (s, 1H), 12.70 (broad s, 2H). Mass spectrum, m/z: 285.2 [M + H]+, 307.1 [M + Na]+.
6a: White moist solid (0.18 g, 8%), 1H NMR (200 MHz, DMSO-d6, δ): 2.58 (s, 3H), 2.92 (t, 2H, J = 7.5 Hz), 3.30 (t, 2H, J = 7.5 Hz), 6.47 (d, 1H, J = 9.0 Hz), 7.18 (m, 1H, J = 4.4 Hz), 7.27 (d, 4H, J = 4.4 Hz), 8.04 (d, 1H, J = 9.0 Hz), 12.89 (broad s, 1H), 14.24 (broad s, 1H). Mass spectrum, m/z: 284.9 [M + H]+, 306.8 [M + Na]+, 322.8 [M + K]+.
2d: White solid (227 mg, 81%), 1H NMR (200 MHz, DMSO-d6, δ): 4.05 (s, 3H), 7.10 (s, 1H), 7.38 (d, 1H), 7.55–7.64 (m, 3H), 8.20–8.32 (m, 3H), 10.54 (s, 1H). 13C NMR (100 MHz, DMSO-d6, δ): 187.8, 176.5, 166.8, 163.2, 155.6, 133.3, 132.6, 131.5, 129.8, 127.1, 117.9, 113.3, 111.5, 107.4, 57.9. Mass spectrum, m/z: 281.3 [M + H]+. Anal. calcd for C17H12O4, %: C 72.85; H 4.32. Found, %: C 72.77; H 4.35.
11: White solid (308 mg, 86%), 1H NMR (500 MHz, DMSO-d6, δ): 3.82 (s, 3H), 5.27 (s, 2H), 7.14 (dd, 1H, J = 8.8 Hz, J = 1.9 Hz), 7.34–7.38 (m, 2H), 7.42 (t, 2H, J = 7.0 Hz), 7.49 (d, 2H, J = 7.6 Hz), 7.56–7.61 (m, 3H), 8.00 (d, 1H, J = 8.8 Hz), 8.01–8.05 (m, 2H). Mass spectrum, m/z: 359.1 [M + H]+, 381.2 [M + Na]+, 397.2 [M + K]+. Anal. calcd for C23H18O4, %: C 77.08; H 5.06. Found, %: C 76.95; H 5.12.
2b: White powder (67%), 1H NMR (500 MHz, DMSO-d6, δ): 7.05 (s, 1H), 7.09 (d, 1H, J = 8.9 Hz), 7.55–7.60 (m, 3H), 8.14 (d, 1H, J = 8.9 Hz), 8.16–8.21 (m, 2H); 10.60 (s, 1H), 12.16 (broad s, 1H). 13C NMR (100 MHz, DMSO-d6, δ): 190.5, 176.3, 167.0, 162.8, 157.1, 133.4, 132.5, 131.5, 129.8, 127.1, 116.8, 116.4, 111.0, 107.8. Mass spectrum, m/z: 267.3 [M + H]+. Anal. calcd for C16H10O4, %: C 72.18; H 3.79. Found, %: C 72.28; H 3.77.
2c: Pale yellow powder (59%), 1H NMR (500 MHz, CDCl3, δ): 3.95 (s, 3H), 7.03 (d, 1H, J = 9.0 Hz), 7.54–7.60 (m, 3H), 8.02–8.07 (m, 2H); 8.40 (d, 1H, J = 9.0 Hz), 10.69 (s, 1H), 12.49 (s, 1H). 13C NMR (100 MHz, DMSO-d6, δ): 192.0, 173.5, 167.6, 157.0, 154.8, 142.0, 135.0, 131.0, 130.4, 128.8, 128.1, 116.8, 116.2, 108.8, 60.3. Mass spectrum, m/z: 297.2 [M + H]+, 319.1 [M + Na]+, 335.0 [M + K]+. Anal. calcd for C17H12O5, %: C 68.92; H 4.08. Found, %: C 68.79; H 4.15.
Conclusions
Two novel types of carbonyl derivatives of flavones with two different proton-transfer sites, able to undergo different types of ESIPT were synthesized. All the hydroxyflavones investigated undergo ESIPT in their electronically excited states. ESIPT involving 3-hydroxyl and 4-carbonyl groups was found to have higher barrier compared to ESIPT involving 7-hydroxyl and 6/8-carbonyl fragments. 3,7-Dihydroxy-2-phenyl-6-(3-phenylpropanoyl)-4H-chromen-4-one (1a) probably undergoes ESIDPT via formation of a transition tautomer T6*. This kind of ESIDPT leads to a tautomeric form with a very low rate constant of radiative deactivation of the excited state, what results in low fluorescence quantum yields. 3,7-Dihydroxy-4-oxo-2-phenyl-4H-chromene-8-carbaldehyde most probably undergoes single ESIPT, however, we hope further investigations will reveal more details on the behavior of this compound.Acknowledgements
The research was financed by the Polish National Science Centre (NCN) under Grant No. 2014/13/N/ST4/04105. PhD studies of I. E. Serdiuk were supported by scholarship of the Polish Bureau for Academic Recognition and International Exchange (BUWiWM). The authors gratefully acknowledge Prof. dr hab. Wiesław Wiczk for valuable consultations and technical support on the low-temperature and time-resolved fluorescence investigations. A special acknowledgment is addressed to Prof. dr hab. inż. Jerzy Błażejowski for technical support.Notes and references
- J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803 RSC.
- J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615 CrossRef CAS PubMed.
- S. Protti and A. Mezzetti, Photochemistry, 2012, 40, 295 CAS.
- C. Randino, M. Ziołek, R. Gelabert, J. A. Organero, M. Gil, M. Moreno, J. M. Lluch and A. Douhal, Phys. Chem. Chem. Phys., 2011, 13, 14960 RSC.
- A. Mordziński, A. Grabowska, W. Kühnle and A. Kröwczyński, Chem. Phys. Lett., 1983, 101, 291 CrossRef.
- R. Wortmann, S. Lebus, H. Reis, A. Grabowska, K. Kownacki and S. Jarosz, Chem. Phys. Lett., 1999, 243, 295 CAS.
- V. Enchev, N. Markova, M. Stoyanova, P. Petrov, M. Rogozherov, N. Kuchukova, I. Timtcheva, V. Monev, S. Angelova and M. Spassova, Chem. Phys. Lett., 2013, 563, 43 CrossRef CAS.
- E. Falkovskaia, V. G. Pivovarenko and J. C. Valle, J. Phys. Chem. A, 2003, 107, 3316 CrossRef CAS.
- A. D. Roshal, V. I. Moroz, V. G. Pivovarenko, A. Wróblewska and J. Błażejowski, J. Org. Chem., 2003, 68, 5860 CrossRef CAS PubMed.
- V. V. Moroz, A. G. Chalyi, I. E. Serdiuk, A. D. Roshal, B. Zadykowicz, V. G. Pivovarenko, A. Wróblewska and J. Błażejowski, J. Phys. Chem. A, 2013, 117, 9156 CrossRef CAS PubMed.
- D. A. Svechkarev, A. O. Doroshenko and D. Y. Kolodezny, Cent. Eur. J. Chem., 2012, 10, 205 CrossRef CAS.
- S. Formosinho and L. G. Arnaut, J. Photochem. Photobiol., A, 1993, 75, 21 CrossRef CAS.
- M. Wera, A. G. Chalyi, A. D. Roshal, B. Zadykowicz and J. Błażejowski, Struct. Chem., 2014, 25, 969 CrossRef CAS.
- G. Geetha, K. Viswanathan and S. N. D. Pradeep, Org. Lett., 2002, 4, 781 CrossRef.
- I. E. Serdiuk, A. D. Roshal and J. Błażejowski, Chem. Heterocycl. Compd., 2014, 50, 396 CrossRef CAS.
- J. C. Duff and E. J. J. Bills, J. Chem. Soc., 1934, 1305 RSC.
- Y. S. Chi, T. T. Dao, H. P. Kim, J. Kim, H. Park and S. Kim, Bioorg. Med. Chem. Lett., 2004, 14, 1165 CrossRef PubMed.
- I. E. Serdiuk, A. S. Varenikov and A. D. Roshal, J. Phys. Chem. A, 2014, 118, 3068 CrossRef CAS PubMed.
- A. S. Varenikov, I. E. Serdiuk and A. D. Roshal, Fr.-Ukr. J. Chem., 2013, 1, 153–157 CAS.
- I. E. Serdiuk, M. Wera, A. D. Roshal and J. Błażejowski, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, 895 Search PubMed.
- O. S. Wolfbeis and R. Schipfer, Berichte der Bunsengesellschaft für physikalische Chemie, 1982, 86, 237 CrossRef CAS.
- G. A. Brucker, T. C. Swinney and D. F. Kelley, J. Phys. Chem., 1991, 95, 3190 CrossRef CAS.
- A. D. Roshal, J. A. Organero and A. Douhal, Chem. Phys. Lett., 2003, 379, 53 CrossRef CAS.
- P. K. Sengupta and M. Kasha, Chem. Phys. Lett., 1979, 68, 382 CrossRef CAS.
- N. A. Tyukavkina and N. N. Pogodaeva, Chem. Nat. Compd., 1971, 7, 8 CrossRef.
- E. Otyepkova, T. Nevěčná, J. Kulhánek and O. Exner, J. Phys. Org. Chem., 2003, 16, 721 CrossRef CAS.
- R. D. B. Fraser and E. Suzuki, in Spectral Analysis, ed. J. A. Blackburn, Marcel Dekker, New York, 1970, p. 171 Search PubMed.
- J. B. Birks, in Photophysics of Aromatic Molecules, ed. J. B. Birks, Wiley, London, 1970, p. 718 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For absorption, phosphorescence, fluorescence 3D and excitation spectra as well as 1H-NMR and mass spectra of new compounds. See DOI: 10.1039/c5ra13912k |
| This journal is © The Royal Society of Chemistry 2015 |