
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogenation of allyl alcohols catalyzed by aqueous palladium and platinum nanoparticles†
Krystel
Di Pietrantonio
a,
Francesca
Coccia‡
a,
Lucia
Tonucci
*b,
Nicola
d'Alessandro
a and
Mario
Bressan
a
aDepartment of Engineering and Geology, G. d'Annunzio University of Chieti-Pescara, Italy
bDepartment of Philosophical, Educational and Economic Sciences, G. d'Annunzio University of Chieti-Pescara, Italy. E-mail: l.tonucci@unich.it; Tel: +39 0871 3555340
First published on 6th August 2015
Abstract
A series of Pd and Pt nanoparticles (NPs) was prepared starting from the corresponding metal ions and lignosulphonates; NPs were tested as catalysts for allyl alcohols hydrogenation in water at room temperature and pressure. All NPs were active with sharp differences in conversions and selectivities: Pt NPs formed mainly saturated alcohols, whereas Pd NPS were more active, but less selective, forming, in addition to saturated alcohols, also isomeric unsaturated alcohols and aldehydes, both saturated and unsaturated.
Introduction
Low molecular weight saturated alcohols are currently studied in order to replace, at least in part, traditional fuels, for their good miscibility with hydrocarbons, high energy content and low vapor pressure.1,2 At present, for one of the most promising derivative, i.e. butyl alcohol, yields for the current biotechnological process and costs are far to be satisfactory.3–5 At the contrary, a useful building block for chemical and pharmaceutical processes, 1-propanol,6 could be obtained by hydrogenation of 2-propen-1-ol, which, in turn, can be produced by deoxydehydration of glycerol, a by-product of biodiesel production, available in large amounts and at relatively low costs.7–9In general, the hydrogenation reactions have a crucial role in the preparation of required products for biorefinery and for the flavor, fragrance and pharmaceutical industries,10–15 which are obtained from highly oxygenated biomasses by lowering their O/H ratio.16 Hydrogenations are normally performed using hydrogen gas and heterogeneous metal catalysts, such as Raney® nickel or palladium, rhodium or platinum metals.17–20 The selectivity in the reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C group in the presence of other functional substituents (e.g., CO or OH), can be controlled by a variety of parameters, such as the nature of the metal catalyst, the presence of a second metal (bimetallic catalysts), the size of metal particles and the nature of the supporting materials (electron-donating or -withdrawing ligand effects, metal–support interactions and solvent effect).21–26
C group in the presence of other functional substituents (e.g., CO or OH), can be controlled by a variety of parameters, such as the nature of the metal catalyst, the presence of a second metal (bimetallic catalysts), the size of metal particles and the nature of the supporting materials (electron-donating or -withdrawing ligand effects, metal–support interactions and solvent effect).21–26
Metal nanoparticles (NPs) are becoming increasingly attractive for industrial catalysis, thanks to their high surface-to-volume ratio, easy preparation and tunable dimensions.27,28 In particular, some palladium and platinum NPs are reported to be active catalysts in the hydrogenation of unsaturated hydrocarbons or allyl alcohols.29–35 However, the preparation of metal NPs often involves the use of environmental unfriendly organic solvents. For this reasons, the principles of green chemistry prompted researchers to prepare new water “soluble” metal NPs, utilizing biochemical methods or, to prevent an extensive agglomeration, natural renewable stabilizers.36–41
In the present study, we investigated the hydrogenation of allyl alcohols catalyzed by Pt and Pd NPs, stabilized by lignins and prepared according to our previously published method.42,43 To reduce and stabilize metal ions we used different types of lignin, an important and widely available waste-product of paper and sugar cane industries, which, at present, is mainly valorized for energetic uses.44,45 In particular, we utilized kraft lignin, lignosulphonates and low-sugar lignosulphonates.
Results and discussion
We prepared a series of twelve NPs using two metals (Pt and Pd) and six water-soluble lignins: kraft lignin (KrLig), ammonium lignosulphonate (AmLig), calcium (CaLig) and sodium (NaLig) lignosulphonates and the corresponding low-sugar ones (CaROLig and NaROLig); lignins, in water solution and at 80 °C, acted as both reducing agents for the metal ions, i.e. H2PtCl6 and PdCl2, and as stabilizing agents for the formed nanoparticles. The formation of zero-valent NPs was signaled by the colour change of the solution (from brownish-yellow to brown-black, with no evidence for solid precipitates) and confirmed by UV-vis spectra (Fig. S1 and S2†); the integrity of the polymers was confirmed by 1H NMR (Fig. S3–S6†) and IR spectra (Fig. S7–S9†).42 The NPs are stable, by UV-vis data, for 30 days at air and at 20 °C.TEM images were collected for all freshly prepared NPs (Fig. 1 and S10–S19†). As reported in Fig. 1, the Pd NPs are spherical while the Pt NPs are mostly irregular in shape and smaller than palladium ones.
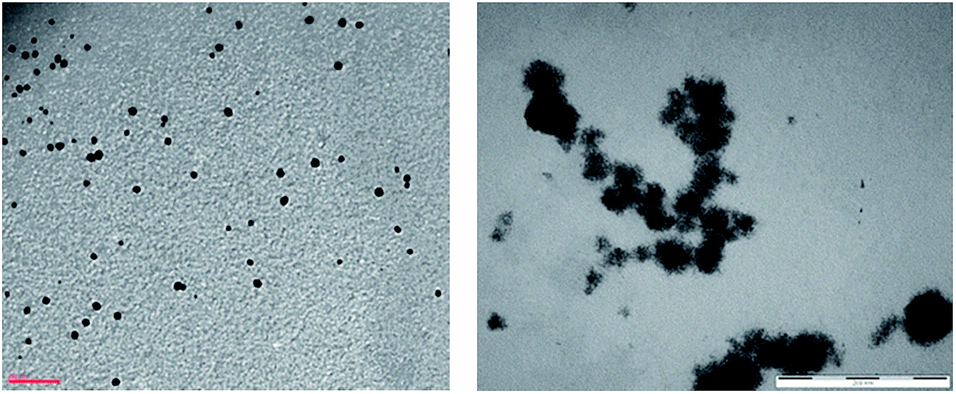 | ||
| Fig. 1 TEM images of CaROLig Pd (left, scale bar = 100 nm) and CaROLig Pt (right, scale bar = 200 nm). | ||
In Table 1 the sizes of NPs are reported, as measured by TEM and DLS. For Pd NPs, the sizes obtained by TEM are always comparable with the results obtained from DLS spectra (except in the single case of AmLig NPs). For Pt NPs, TEM images did not allow accurate measurement of the particle size, because of their irregular shape and also because of the diffuse formation of clusters. Apparently, different lignins gave rise to nanoparticles with different sizes; in particular CaLig and NaLig NPs resulted invariably larger than nanoparticles obtained in presence of the corresponding low-sugar lignins, CaROLig and NaROLig. However, if one looks only to the largely spherical and regular Pd NPs, the measured diameters always fall in a relatively narrow range, 19.2 nm ± 6.8 (by TEM) and 15.8 nm ± 7.5 (by DLS), thus suggesting quite similar behaviours for the various lignins employed.
| Lignin | Diameter of M NPs (nm) | ||
|---|---|---|---|
| by TEM | by DLS | ||
| M = Pd | M = Pd | M = Pt | |
| NaROLig | 10 | 11 | 106 |
| KrLig | 12 | 9 | 5 |
| CaROLig | 15 | 17 | 31 |
| NaLig | 24 | 26 | 58 |
| CaLig | 26 | 27 | 52 |
| AmLig | 28 | 5 | 132 |
The XRD analysis of dried powders indicated a crystalline structure of all NPs and the peaks are identical to those previously reported for Pd and Pt zero-valent nanoparticles (Fig. S20 and S21†).46,47
We conducted first the hydrogenation of 2-propen-1-ol by hydrogen gas at room pressure, in presence of the above Pd and Pt NPs, in water solution at 20 °C. Then, to evaluate the behaviour of catalysts with increasing substitution at C-sp2 atom and the carbon chain extension, we performed the hydrogenation of substituted water-soluble allyl alcohols, i.e. 2-buten-1-ol, 3-methyl-2-buten-1-ol, cis-2-penten-1-ol, trans-2-penten-1-ol. In presence of metal catalysts and hydrogen gas, several reactions are possible, i.e. CC reduction (Scheme 1, reaction a), isomerization to saturated aldehydes (Scheme 1, reaction b),48–50 hydrodeoxygenation to hydrocarbons (Scheme 1, reaction c),51–53 oxidative dehydrogenation to unsaturated aldehydes (Scheme 1, reaction d)54–56 and isomerization by double-bond migration (Scheme 1, reaction e).57,58
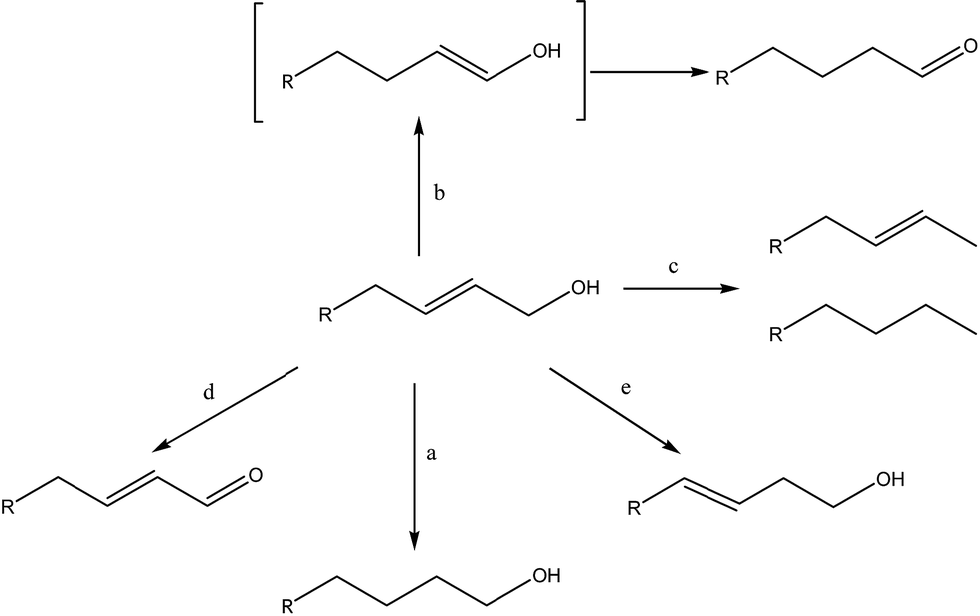 | ||
| Scheme 1 Reactivity of an allyl alcohol in presence of H2 gas and metal catalysts. | ||
The catalytic activity of our NPs was tested in presence of 2-propen-1-ol (20 mM) as the model substrate: the reactions were carried out for 24 hours in presence of 0.04 mmol L−1 Pd NPs or 0.07 mmol L−1 Pt NPs and hydrogen gas at 1.013 barr in water. At preset time intervals we evaluated by 1H NMR spectra the course of hydrogenation. The temperature of reaction was defined at 20 °C; at 5 °C the reactions were definitely too slow, whereas at higher temperatures (i.e., 40 °C) no better conversions and selectivities were obtained.
Alcohol conversions were definitely faster when Pd NPs were fluxed in water with H2 gas for 20 minutes, before adding the organic substrate. The above prolonged H2 flux had no effect in presence of Pt NPs. Finally, we performed the reactions in 100% D2O, but no incorporation of deuterium in the hydrogenated products was observed. In the absence of metal NPs, all substrates were quantitatively recovered unchanged, whereas, when the reactions were conducted in presence of 60 mg of lignins, we observed small and erratic amounts of hydrogenation products, always below 5% molar yield.
Platinum catalysts
During the hydrogenation of 2-propen-1-ol by Pt NPs, conversions were generally poor, below 40%, apart the single case of Pt NaLig NPs, which exhibited almost quantitative conversion (Table 2).| Catalyst | Substrate | ||||
|---|---|---|---|---|---|
| 2-Propen-1-ol | 2-Buten-1-ol | 3-Methyl-2-buten-1-ol | trans-2-Penten-1-ol | cis-2-Penten-1-ol | |
| a Conversions, % (yields, %: saturated alcohol, 2-unsaturated aldehyde, alkane/alkene), at 24 h. Reaction conditions, see Experimental. Data by 1H NMR and GC. Values are mean of three replicates; percent errors are always <10%. | |||||
| Pt CaLig NPs | 18 (16, 0, n.d.) | 22 (21, 0, n.d.) | 31 (12, 7, n.d.) | 30 (21, 2, 6) | 100 (75, 0, 10) |
| Pt CaROLig NPs | 34 (10, 0, n.d.) | 49 (24, 7, n.d.) | 30 (10, 7, n.d.) | 45 (22, 0, 9) | 45 (36, 0, 8) |
| Pt AmLig NPs | 30 (24, 0, n.d.) | 40 (18, 6, n.d.) | 36 (10, 5, n.d.) | 52 (31, 0, 10) | 45 (36, 0, 9) |
| Pt KrLig NPs | 39 (16, 0, n.d.) | 45 (17, 8, n.d.) | 38 (6, 11, n.d.) | 33 (17, 6, 9) | 39 (36, 0, 5) |
| Pt NaLig NPs | 87 (56, 0, n.d.) | 46 (20, 6, n.d.) | 36 (7, 8, n.d.) | 40 (25, 8, 7) | 64 (63, 0, 6) |
| Pt NaROLig NPs | 65 (60, 0, n.d.) | 25 (15, 8, n.d.) | 17 (11, 4, n.d.) | 33 (19, 8, 6) | 60 (37, 0, 12) |
Commercial Pt/C gave, in the same reaction conditions, quantitative conversion of 2-propen-1-ol, leading to 1-propanol (70%) and to significant amounts of the saturated aldehyde propanal. By contrast, in the reaction catalyzed by the Pt NPs, the only product found in the aqueous liquid phase was 1-propanol (even if with yields highly dependent upon the nature of the catalyst, from 10% to 60%), with no evidence for the products of isomerization (propanal) or of oxidative dehydrogenation (propenal). Clearly, there was a serious gap between the amount of 1-propanol formed and the amount of 2-propen-1-ol reacted. In fact, when the headspace of the reaction vessels was analyzed by GC-MS, light hydrocarbons, i.e. propene (m/z 42) and propane (m/z 44), were detected (Fig. 2); to confirm this unexpected result, we synthesized deuterated 2-propen-1-ol-d5, which upon hydrogenation in the presence of Pt NaLig NPs gave rise to propene-d5 (m/z 47) and propane-d5 (m/z 49), detected by GC-MS in the headspace (Fig. 3).
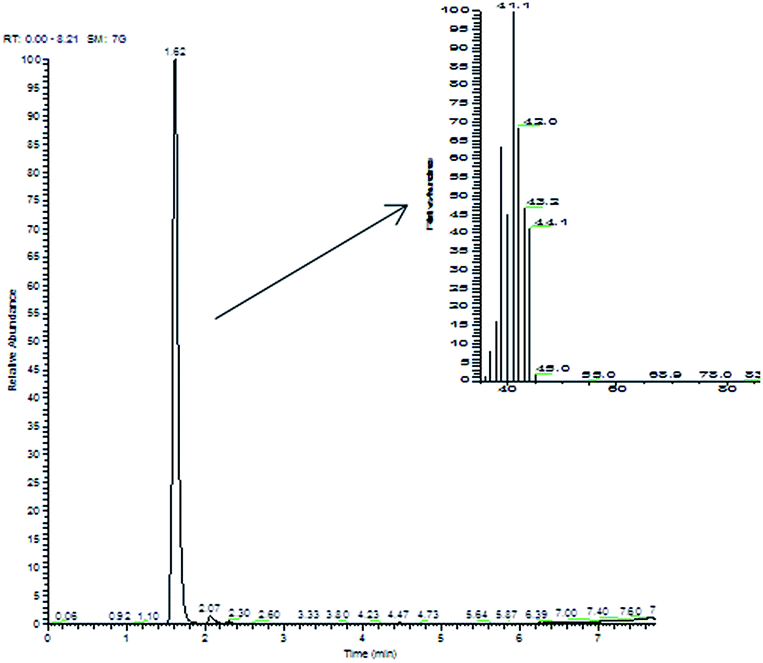 | ||
| Fig. 2 Chromatogram and mass spectrum of headspace of 2-propen-1-ol hydrogenation by Pt NaLig NPs. | ||
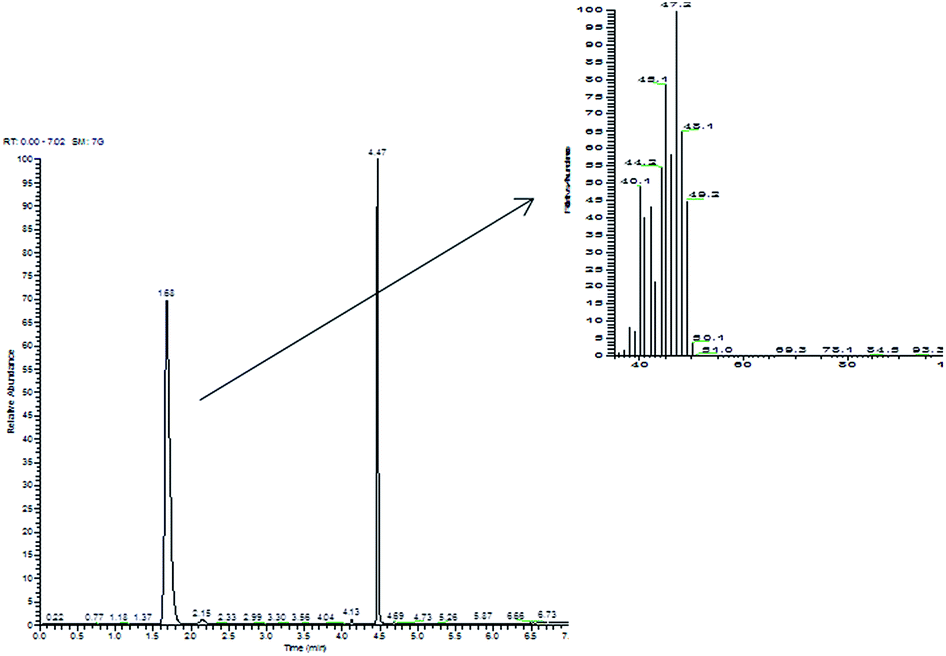 | ||
| Fig. 3 Chromatogram and mass spectrum of headspace of 2-propen-1-ol-d5 hydrogenation by Pt NaLig NPs. The signal at 4.47 is due to an impurity from the synthesis of 2-propen-1-ol-d5. | ||
It should also be noted that the reaction rate with Pt NPs (in particular with Pt NaROLig NPs), while comparable to that of Pt/C within the first times of the reaction, significantly slowed down becoming practically zero in a few hours (Fig. 4). This strongly suggests a significant loss of activity during the reaction: indeed, TEM images showed important changes in the morphology of Pt NPs during the hydrogenation, the NPs underwent extensive aggregation to highly branched and irregular species, possibly responsible of the overall poor performance of the Pt nanocatalysts (Fig. 5).
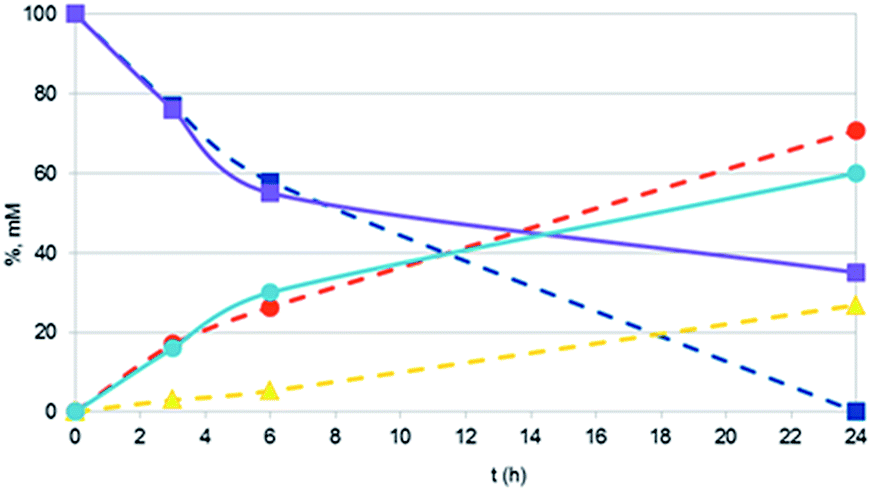 | ||
| Fig. 4 Time course of 2-propen-1-ol hydrogenation by Pt/C (dashed lines) and Pt NaROLig NPs (solid lines): 2-propen-1-ol (squares); 1-propanol (circles); propanal (triangles). Reaction conditions, see Experimental. Data by 1H NMR. | ||
 | ||
| Fig. 5 TEM images of Pt AmLig NPs before (left, scale bar = 50 nm) and after the hydrogenation reaction (right, scale bar = 100 nm). | ||
Conversions of 2-buten-1-ol and 3-methyl-2-buten-1-ol (cis/trans mix) were again rather poor, always below 50%, with very low yields in the corresponding saturated alcohols; moreover, and in contrast with 2-propen-1-ol, minor amounts of the oxidative dehydrogenation products, i.e. 2-butenal or 3-methyl-2-butenal, were detected. Also in this cases, saturated aldehydes were never observed, whereas the corresponding light hydrocarbons, i.e. butane/2-butene or 2-methylbutane/2-methylbutene, were detected by the GC-MS of headspace, although no reliable quantification was possible.
Also trans-2-penten-1-ol was poorly transformed (30–52%), the pivotal product being 1-pentanol, together with minor amounts of 2-pentenal (up to 8%) and of the corresponding hydrocarbons pentane and/or pentene, which, in this case, could be reliably quantified by flushing the reaction mixtures into a chloroform trap and then analyzing by GC. Mass balances could be satisfactorily closed in almost all experiments. Conversions of cis-2-penten-1-ol were definitely larger than those of the trans-isomer (between 39% and 100%) and with 1-pentanol and pentane and/or pentene as the only products detected, with yields in 1-pentanol as high as 75%. Moreover, 1-pentanol appears to favoured in the case of the cis-isomer, which strongly suggests a syn addition of hydrogen to the double bond, more sensitive to steric hindrance of the substituents (the same trend was observed also in the case of the Pd-catalyzed reactions, vide infra) Table 3.
| Catalyst | Substrate | ||||
|---|---|---|---|---|---|
| 2-Propen-1-ol | 2-Buten-1-ol | 3-Methyl-2-buten-1-ol | trans-2-Penten-1-ol | cis-2-Penten-1-ol | |
| a Conversions, % (yields, %: saturated alcohol, 2-unsaturated aldehyde, saturated aldehyde, 3-unsaturated alcohol), at 24 h. Reaction conditions, see Experimental. Data by 1H NMR and GC. Values are mean of three replicates; percent errors are always <10%. | |||||
| Pd CaLig NPs | 100 (22, 0, 74) | 38 (21, 8, 9, 0) | 42 (12, 13, 0, 17) | 87 (37, 0, 8, 33) | 85 (37, 0, 11, 37) |
| Pd CaROLig NPs | 100 (28, 0, 72) | 80 (24, 7, 21, 0) | 37 (4, 14, 0, 18) | 86 (48, 0, 6, 27) | 92 (58, 0, 7, 18) |
| Pd AmLig NPs | 100 (24, 0, 66) | 80 (18, 0, 20, 0) | 43 (6, 5, 0, 10) | 95 (42, 0, 8, 23) | 100 (58, 0, 13, 27) |
| Pd KrLig NPs | 100 (39, 0, 55) | 50 (17, 6, 12, 0) | 54 (15, 7, 0, 18) | 74 (39, 0, 8, 28) | 95 (49, 0, 11, 20) |
| Pd NaLig NPs | 55 (16, 0, 39) | 23 (20, 0, 0, 0) | 51 (21, 7, 0, 10) | 100 (51, 0, 11, 20) | 40 (17, 0, 10, 10) |
| Pd NaROLig NPs | 100 (24, 0, 63) | 77 (15, 16, 16, 0) | 47 (9, 18, 0, 16) | 91 (55, 0, 8, 28) | 100 (87, 0, 7, 0) |
Palladium catalysts
In presence of Pd NPs, conversions of 2-propen-1-ol were always quantitative (apart with Pd NaLig NPs; Table 3). However, the only products detected were always propanal (up to 74%) and 1-propanol (up to 39%), with no evidence for propenal or propane/propene. In all examined cases, mass balances were quite satisfactory.Quantitative conversions and comparable yields of 1-propanol (29%) and propanal (71%) were also found in the reaction conducted in the same conditions with commercial Pd/C. However, by following the time courses of the hydrogenations, we found that the catalytic activity of the nanocatalyst (namely Pd CaROLig NPs) was much larger than that of Pd/C (Fig. 6), so that the reaction with Pd NPs reached quantitative conversion of 2-propen-1-ol within few hours. TEM images of the reaction mixture showed that the Pd NPs, while remaining spherical during the reaction, underwent extensive disaggregation, giving rise to many very small particles – less than 7 nm in diameter, to be compared with the 15–17 nm size of the initial NPs (Fig. 7) – possibly responsible for the observed enhanced reactivity of the nanocatalyst.
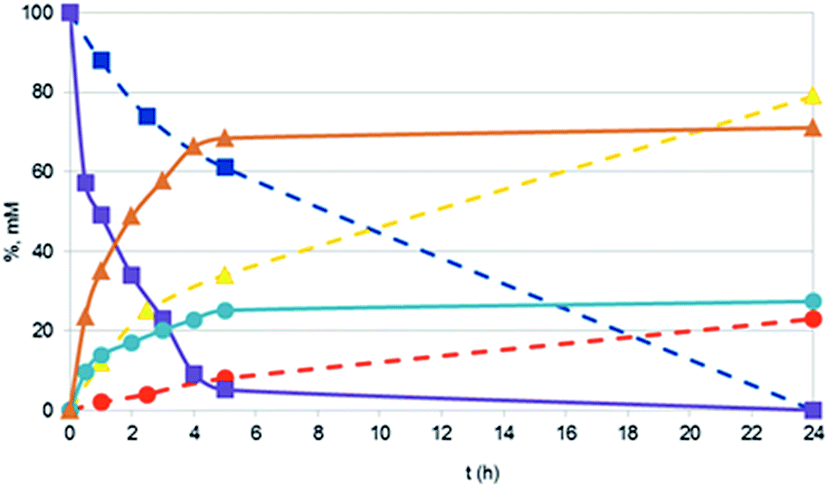 | ||
| Fig. 6 Time course of 2-propen-1-ol hydrogenation by Pd/C (dashed lines) and Pd CaROLig NPs (solid lines): 2-propen-1-ol (squares); 1-propanol (circles); propanal (triangles). Reaction conditions, see Experimental. Data by 1H NMR. | ||
 | ||
| Fig. 7 TEM images of Pd AmLig NPs before (left, scale bar = 100 nm) and after the hydrogenation reaction (right, scale bar = 100 nm). | ||
In the hydrogenation of 2-buten-1-ol and 3-methyl-2-buten-1-ol (cis/trans mix), conversions were strongly dependent upon the nature of the lignin employed, ranging from 23% to 80% and also selectivity was rather erratic, with the formation of all the possible products, i.e. of double bond hydrogenation (1-butanol or 3-methylbutan-1-ol), of oxidative dehydrogenation (2-butenal or 3-methyl-2-butenal, up to 18%; it should be noted that these products could not be detected in the first 6 hours of reaction), of isomerization (butanal, up to 21%; 3-methylbutanal was not detected) and also of double bond migration (3-methyl-3-butenol, up to 18%; 3-butenol was not detected). Light hydrocarbons were never detected, nevertheless overall mass balances were largely unsatisfactory.
By contrast, the conversions of 2-penten-1-ols (both cis- and trans- isomers) were rather good, almost always quantitative, with 1-pentanol as the major product. Whereas pentenal was always absent and pentanal below 13%, significant amounts of 3-penten-1-ol, arising upon double bond migration, were observed (10–37%). Overall mass balances were generally satisfactory.
The isomerization pathway to the saturated aldehyde by Pd NPs was prevalent only for 2-propen-1-ol and less significant for the other examined substrates, in agreement with literature data reporting that the presence of C-sp2 substituents (methyl, ethyl, dimethyl versus hydrogen, in our case) generally depresses the selectivity toward carbonyl products,58 since the mechanism of the isomerization implies an hydrogen insertion into the terminal C-sp2 and the formation of an enol, which tautomerizes to the corresponding carbonyl.50 Although isomerization of unsaturated alcohols to carbonyl derivatives is generally considered a minor reaction, in recent years highly selective processes have begun to attract growing interest.59,60 Conversely, also the high yields of 1-pentanol from trans-2-penten-1-ol could be of interest, in view of the poorer selectivities reported in the literature.58
Conclusions
All the examined metal NPs act as hydrogenation catalysts for allyl alcohols, albeit with very different yields and selectivities.Broadly speaking, Pd NPs are far more catalytically active than the corresponding platinum-derivatives, but definitely favour the formation of saturated aldehydes over unsaturated alcohols, in line with the well-known reactivity of the conventional heterogeneous Pd/C catalysts.
However, there are definite evidence that, during the reaction, the structure of the metal cores significantly modifies, giving rise to significant disaggregation (in the case of Pd NPs) or aggregation (in the case of Pt NPs) of the nanoparticles and therefore to more or less reactive species, respectively. In fact, Pd NPs, after the addition of fresh amounts of the substrate (i.e., 2-propen-1-ol) into the 24 h reaction mixture, continued to catalyze the reactions with same selectivity (although at a lower rate, 25% of conversion after 24 h). In this case, it is possible that a structural change of lignin takes place, resulting in slowing down the passage of the substrate throughout the polymeric “protective shield” of the definitively smaller (and in principle more active) nanoparticles. Conversely, by adding fresh substrate into the Pt NPs reaction mixture, the reaction did not restart at all, likely because the observed extensive aggregation of NPs (Fig. 5) reduces the available metal surface area.
The nature of the lignins employed to stabilize the nanoparticles does not result in the formation of metal-cores very different in size; nevertheless, different lignins resulted in different reactivities of the obtained NPs, for both metals, although in a very obscure way. At the moment, we can only speculate that each one of the lignins employed (exhibiting different elemental compositions, in particular the C/O ratios), may differ in polarity, degree of cross-linking and swallowability toward water, thus influencing the capability of the substrates to reach more or less easily the metal core.
Conversions of the various allyl alcohols, for both Pd and Pt NPs, generally follow the Lebedev rule, which establishes that the number of substituents attached to C-sp2 atoms depresses the reactivity of the substrate versus hydrogen:61 indeed, the least substituted and least hindered substrate, i.e. 2-propen-1-ol, is the most reactive and, generally speaking, the reactivity decreases from 2-propen-1-ol to 2-buten-1-ol and to 3-methyl-2-buten-1-ol. However, 2-penten-1-ols exhibit a reactivity similar to the short-chain 2-propen-1-ol. Finally, Pd NPs were less selective than Pt NPs for the corresponding saturated alcohols, although with notable exceptions.
Although yields and conversions are not always satisfactory, when compared to those of the conventional heterogeneous catalysts, this study could be an important step aiming to design a “one-pot” reaction from glycerol, the natural feedstock from diesel production plants, to 1-propanol, an important commodity chemical. A preliminary test showed that: by purging continuously with hydrogen a 2,4-dimethyl-3-pentanol solution of glycerol and methyltrioxorhenium catalyst, collecting the gas phase (containing 2-propen-1-ol) in a cold water trap in the presence of catalytic amounts of Pt NaROLig NPs, 1-propanol can be obtained with high selectivity.
Experimental
Materials
Calcium lignosulphonates (CaLig), ammonium lignosulphonates (AmLig), sodium lignosulphonates (NaLig), calcium and sodium low-sugar lignosulphonates (CaRO and NaRO) were a gift from Burgo Group S.p.A. (Tolmezzo, Italy). Kraft lignin (KrLig) was purchased from Sigma-Aldrich. Chloroplatinic acid (H2PtCl6·6H2O), and palladium chloride (PdCl2) were purchased from Strem Chemicals. Deuterated 2-propen-1-ol-d5 was prepared by literature.7 All the other remaining reagents were bought from Sigma-Aldrich. C, H, N and S elemental analyses of lignins were carried out by the 1106 Carlo-Erba Elemental Analysis (see Table 4).| %C | %H | %N | %S | |
|---|---|---|---|---|
| KrLig | 61.47 | 5.71 | 0.68 | 1.47 |
| NaLig | 45.71 | 4.64 | 0.12 | 4.02 |
| CaROLig | 38.99 | 5.03 | 0.14 | 6.06 |
| AmLig | 41.72 | 5.19 | 3.57 | 7.48 |
| NaROLig | 39.89 | 5.00 | 0.15 | 5.88 |
| CaLig | 36.55 | 4.26 | 0.16 | 5.29 |
Synthesis and characterization of metal NPs
Lignin stabilized Pd and Pt nanoparticles were prepared in accordance with our previous work.42 In brief, 0.06 g lignin were added to 0.03 g H2PtCl6·6H2O in 6 mL water or to 0.01 g PdCl2 in 10 mL water. The solution was heated to 80 °C for 3 h. The obtained NPs solution was used as such in the hydrogenation experiments.The nanoparticles were fully characterized with TEM, DLS, UV-vis, XRD and FT-IR. The UV-vis analyses were carried out in 1 cm quartz cuvettes (Hellma) by diluting 50 μL of the NPs solution in 10 mL (for Pd) or 20 mL (for Pt) of ultrapure water. The UV-vis instrument was a Jenway 6505 spectrophotometer; with a spectral window range of 200 nm to 600 nm.
The samples for Fourier transform infrared (FT-IR) spectroscopy were prepared by evaporating off the water solvent from the NPs. The resulting solid residue was milled with KBr (1%, w/w) to form a very fine powder that was compressed into a thin pellet and analyzed using the FT-IR instrument (Perkin Elmer 1600), using a spectral window from 4000 cm−1 to 400 cm−1.
The samples for the TEM measurements were prepared by diluting 100 μL of NPs solution in 10 mL water and placing it onto 3 mm, 300 mesh, formvar/carbon nickel grids (Agar Scientific Ltd), which were left drying in air at room temperature for 48 h to permit the evaporation of the solvent. The TEM measurements were performed under vacuum using a 109 Zeiss EM equipped with built-in electromagnetic objective lenses and a camera (Oberkochen, Germany).
For the DLS analyses, the samples were prepared by adding 200 μL of the NPs solution in 1.8 mL of ultrapure water and put in a quartz cell for the acquisition. Experiments were performed on a Zetasizer Nano ZS, Malvern Instrument Ltd., U.K.
The samples for XRD analysis were prepared by solvent evaporation with rotary evaporator at 30 °C of the 10 mL solution of Pd and Pt NPs and the resulting solid powder was placed on the glass plate of the instrument. The XRD analysis was performed on a Miniflex II Rigaku automated power XRD system (Cu Kα radiation, 45 kV, 100 mA) (RINT 2500, Japan). Diffraction data were recorded using continuous scanning at 3° min−1, with 0.010° steps and a PDF-4/mineral 2013 database.
GC analyses were performed on an HP 68970 GLC instrument equipped with a FID, using a 30 m HP-5 capillary column (0.32 mm i.d.; 0.25 film thick) with the injection port kept at 250 °C (carrier gas: He).
The gas chromatography-mass spectroscopy (GC-MS) apparatus comprised a Thermo Scientific Focus series gas-chromatograph coupled to an ISQ mass-selective detector equipped with a split/splitless injection system (injections made in split mode) and an HP-5 MS (cross-linked 5% phenyl methyl siloxane) capillary column (30 m length, 0.25 mm diameter, 0.1 μm film thickness) using helium as carrier gas at constant pressure of 30 kPa. The acquisition parameters were: source at 250 °C, transfer line at 250 °C, 3 min of delay acquisition time at the beginning of the analysis, mass range 33–350 amu, injector temperature 250 °C, initial temperature of the analyses 60 °C (1 min), then 10 °C min−1 up to 250 °C (kept for 6 min), for a total acquisition time of 26 min. The sample injected was 1 μL.
NMR spectra were obtained using a Bruker Avance 300 spectrometer (7.05 Tesla) equipped with a high resolution multinuclear probe that operated in the range of 30 MHz to 300 MHz. To eliminate the dominant water signal in the 1H NMR spectra, water suppression was carried out using a presaturation sequence of a composite pulse (zgcppr Bruker sequence). A co-axial capillary tube that contained a 30 mM D2O solution of 3-(trimethylsilyl) propionic-2,2,3,3-d4 acid, sodium salt, was used as the reference.
Hydrogenation procedure
15 mL of deionized water was placed into an hermetically closed 20 mL vial and then 100 μL of Pd NPs (0.56 μmol Pd; 0.04 mmol Pd/L solution) or 100 μL of Pt NPs (0.97 μmol Pt; 0.07 mmol Pt/L solution) solution were introduced; the mixture was fluxed bubbling hydrogen gas for 20 minutes. The substrate (0.3 mmol; 20 mM) was added. The reaction solution was left under static H2 atmosphere at room pressure (1.013 barr) and temperature (20 °C) and stirred for 24 h.In the reactions catalyzed with commercial Pt/C and Pd/C, we used 1 mg of 10% Pd/C (0.9 μmol; 0.06 mmol Pd/L solution) or 4 mg of 5% Pt/C (1 μmol; 0.07 mmol Pt/L solution) in 15 mL of water and in presence of 0.3 mmol of allyl alcohol (20 mM).
In a preliminary test, a 2,4-dimethyl-3-pentanol solution (5 mL) of glycerol (5 mmol) and methyltrioxorhenium catalyst (0.1 mmol) held at 140 °C was purged continuously with hydrogen and the gas phase, which contained 2-propen-1-ol, was collected in a cold water trap (100 mL) in the presence of Pt NaROLig NPs catalyst (5 μmol of Pt), producing 1-propanol.
Acknowledgements
We thank “Consorzio di Ricerca per l'Innovazione Tecnologica, la Qualità e la Sicurezza degli Alimenti S.c.r.l.” (CIPE funding 20.12.04; DM 28497) and the “Ministero dell'Istruzione, dell'Università e della Ricerca” (MIUR) for financial support.References
- M. Lapuerta, J. Rodríguez-Fernandez, R. García-Contreras and M. Bogarra, Fuel, 2015, 139, 171 CrossRef CAS.
- W. Tutak, K. Lukács, S. Szwaja and Á. Bereczky, Fuel, 2015, 154, 196 CrossRef CAS.
- Y. Gu, Y. Jiang, H. Wu, X. Liu, Z. Li, J. Li, H. Xiao, Z. Shen, H. Dong, Y. Yang, Y. Li, W. Jiang and S. Yang, Biotechnol. J., 2011, 6, 1348 CrossRef CAS PubMed.
- E. N. Efremenko, N. A. Stepanov, A. B. Nikolskaya, O. V. Senko, O. V. Spiricheva and S. D. Varfolomeev, Catal. Ind., 2011, 3, 41 CrossRef.
- K. Liu, H. K. Atiyeh, O. Pardo-Planas, T. C. Ezeji, V. Ujor, J. C. Overton, K. Berning, M. R. Wilkins and R. S. Tanner, Bioresour. Technol., 2015, 189, 292 CrossRef CAS PubMed.
- S. Atsumi, T. Hanai and J. C. Liao, Nature, 2008, 451, 86 CrossRef CAS PubMed.
- V. Canale, L. Tonucci, M. Bressan and N. d'Alessandro, Catal. Sci. Technol., 2014, 4, 3697 CAS.
- D. Taher, M. E. Thibault, D. Di Mondo, M. Jennings and M. Schlaf, Chem.–Eur. J., 2009, 15, 10132 CrossRef CAS PubMed.
- M. E. Thibault, D. V. DiMondo, M. Jennings, P. V. Abdelnur, M. N. Eberlin and M. Schlaf, Green Chem., 2011, 13, 357 RSC.
- M. Steffan, F. Klasovsky, J. Arras, C. Roth, J. Radnik, H. Hofmeister and P. Claus, Adv. Synth. Catal., 2008, 350, 1337 CrossRef CAS.
- S. Di Dio, M. Marchetti, S. Paganelli and O. Piccolo, Appl. Catal., A, 2011, 399, 205 CrossRef CAS.
- S. De, B. Saha and R. Luque, Bioresour. Technol., 2015, 178, 108 CrossRef CAS PubMed.
- A. B. Bindwal and P. D. Vaidya, Energy Fuels, 2014, 28, 3357 CrossRef CAS.
- E. Falabella Sousa-Aguiar, L. Gorenstin Appel, P. Costa Zonetti, A. do Couto Fraga, A. Azevedo Bicudo and I. Fonseca, Catal. Today, 2014, 234, 13 CrossRef.
- R. Ciriminna, V. Pandarus, F. Béland and M. Pagliaro, Org. Process Res. Dev., 2014, 18, 1110 CrossRef CAS.
- E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gärtner and J. A. Dumesic, Science, 2008, 322, 417 CrossRef CAS PubMed.
- H. Oyamada, T. Naito, S. Miyamoto, R. Akiyama, H. Hagioa and S. Kobayashi, Org. Biomol. Chem., 2008, 6, 61 CAS.
- A. F. Barrero, E. J. Alvarez-Manzaneda, R. Chahboun and R. Meneses, Synlett, 1999, 10, 1663 CrossRef.
- R. A. W. Johnstone, J.-Y. Liu, L. Lu and D. Whittaker, J. Mol. Catal. A: Chem., 2003, 191, 289 CrossRef CAS.
- L. Huang, P. Luo, W. Pei, X. Liu, Y. Wang, J. Wang, W. Xing and J. Huang, Adv. Synth. Catal., 2012, 354, 2689 CrossRef CAS.
- X. Xu, H. Li and Y. Wang, ChemCatChem, 2014, 6, 3328 CrossRef CAS.
- P. Claus, Top. Catal., 1998, 5, 51 CrossRef CAS.
- P. Mäki-Arvela, J. Hájek, T. Salmi and D. Yu Murzin, Appl. Catal., A, 2005, 292, 1 CrossRef.
- P. Claus and Y. Önal, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Weinheim, 2008 Search PubMed.
- V. Ponec, Appl. Catal., A, 1997, 149, 27 CrossRef CAS.
- P. Gallezot and D. Richard, Catal. Rev.: Sci. Eng., 1998, 40, 81 CAS.
- Y. Dai, Y. Wang, B. Liu and Y. Yang, Small, 2015, 11, 268 CrossRef CAS PubMed.
- Z. Guan, S. Lu and C. Li, J. Catal., 2014, 311, 1 CrossRef CAS.
- S. Mandal, D. Roy, R. V. Chaudhari and M. Sastry, Chem. Mater., 2004, 16, 3714 CrossRef CAS.
- S. Bhattacharjee and M. L. Bruening, Langmuir, 2008, 24, 2916 CrossRef CAS PubMed.
- J. Liu, X. Liao and B. Shi, Res. Chem. Intermed., 2014, 40, 249 CrossRef CAS.
- R. Bhandari, D. B. Pacardo, N. M. Bedford, R. R. Naik and M. R. Knecht, J. Phys. Chem. C, 2013, 117, 18053 CAS.
- D. J. Gavia, M. S. Maung and Y.-S. Shon, ACS Appl. Mater. Interfaces, 2013, 5, 12432 CAS.
- Y. Jiang and Q. Gao, J. Am. Chem. Soc., 2006, 128, 716 CrossRef CAS PubMed.
- C. Ornelas, J. R. Aranzaes, L. Salmon and D. Astruc, Chem.–Eur. J., 2008, 14, 50 CrossRef CAS PubMed.
- F. Iram, M. S. Iqbal, M. M. Athar, M. Z. Saeed, A. Yasmeen and R. Ahmad, Carbohydr. Polym., 2014, 104, 29 CrossRef CAS PubMed.
- M. Sayeed Akhtar, J. Panwar and Y.-S. Yun, ACS Sustainable Chem. Eng., 2013, 1, 591 CrossRef.
- W. Chen, L. Zhong, X. Peng, J. Lin and R. Sun, Cellulose, 2014, 21, 125 CrossRef CAS.
- M. Khan, M. Khan, M. Kuniyil, S. F. Adil, A. Al-Warthan, H. Z. Alkhathlan, W. Tremel, M. N. Tahir and M. R. H. Siddiqui, Dalton Trans., 2014, 43, 9026 RSC.
- J. Mittal, A. Batra, A. Singh and M. M. Sharma, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2014, 5, 043002 CrossRef.
- M. J. Rak, T. Friščić and A. Moores, Faraday Discuss., 2014, 170, 155 RSC.
- F. Coccia, L. Tonucci, D. Bosco, M. Bressan and N. d'Alessandro, Green Chem., 2012, 14, 1073 RSC.
- F. Coccia, L. Tonucci, N. d'Alessandro, P. D'Ambrosio and M. Bressan, Inorg. Chim. Acta, 2013, 399, 12 CrossRef CAS.
- S. K. Maity, Renewable Sustainable Energy Rev., 2015, 43, 1427 CrossRef CAS.
- S. K. Maity, Renewable Sustainable Energy Rev., 2015, 43, 1446 CrossRef CAS.
- S. Navaladian, B. Viswanathan, T. K. Varadarajan and R. P. Viswanath, Nanoscale Res. Lett., 2009, 4, 181 CrossRef CAS PubMed.
- H. Tong, H.-L. Li and X.-G. Zhang, Carbon, 2007, 45, 2424 CrossRef CAS.
- D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 2746 CrossRef CAS PubMed.
- M. G. Musolino, C. V. Caia, F. Mauriello and R. Pietropaolo, Appl. Catal., A, 2010, 390, 141 CrossRef CAS.
- E. Sadeghmoghaddam, K. Gaïeb and Y.-S. Shon, Appl. Catal., A, 2011, 405, 137 CrossRef CAS.
- R. Hubaut and J. P. Bonnelle, React. Kinet. Catal. Lett., 1992, 47, 73 CrossRef CAS.
- C. Hoang-Van and O. Zegaoui, Appl. Catal., A, 1997, 164, 91 CrossRef CAS.
- H. Wei, C. Gomez, J. Liu, N. Guo, T. Wu, R. Lobo-Lapidus, C. L. Marshall, J. T. Miller and R. J. Meyer, J. Catal., 2013, 298, 18 CrossRef CAS.
- M. Imachi, N. W. Cant and R. L. Kuczkowski, J. Catal., 1982, 75, 404 CrossRef CAS.
- C. Keresszegi, T. Mallat and A. Baiker, New J. Chem., 2001, 25, 1163 RSC.
- J. Guerra, J. L. Burt, D. A. Ferrer, S. Mejía and M. José-Yacamán, J. Nanopart. Res., 2011, 13, 1723 CrossRef CAS.
- H. Pines, The Chemistry of Catalytic Hydrocarbon Conversions, Academic Press, Inc., New York, USA, 1981 Search PubMed.
- M. G. Musolino, P. De Maio, A. Donato and R. Pietropaolo, J. Mol. Catal. A: Chem., 2004, 208, 219 CrossRef CAS.
- S. Gargiulo, D. J. Opperman, U. Hanefeld, I. W. C. E. Arends and F. Hollmann, Chem. Commun., 2012, 48, 6630 RSC.
- M. Moreno, L. N. Kissell, J. B. Jasinski and F. P. Zamborini, ACS Catal., 2012, 2, 2602 CrossRef CAS.
- S. V. Lebedev, G. G. Kobliansky and A. O. Yakubchik, J. Chem. Soc., Trans., 1925, 127, 417 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c5ra13840j |
| ‡ Present location: Department of Chemistry, University of Milan, Italy. |
| This journal is © The Royal Society of Chemistry 2015 |