
Electrochemical reduction of carbon dioxide to formate with a Sn cathode and an IrxSnyRuzO2/Ti anode
 *a
*a
Abstract
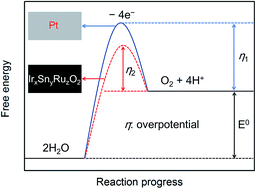
The electrochemical reduction of CO2 to formate has been studied extensively, and most studies have focused on the faradaic efficiency for producing formate. However, the energy efficiency is also critical for the possible industrialization of the process, as it specifies the energy recovered in the product. Here, we report that the energy efficiency is 35.6% when a Pt electrode is used as the anode, and it can increase to 42.1% when an IrxSnyRuzO2/Ti electrode with lower overpotential for oxygen evolution reaction is used as the anode, in spite of their faradaic efficiencies for producing formate are very close (85.1% and 84.8%). When the IrxSnyRuzO2/Ti electrode coated with Nafion membrane is used as the anode, the faradaic efficiency can maintain at 79.6% through a long time electrolysis of CO2 for 500 C.
 Please wait while we load your content...
Please wait while we load your content...

