
Poly(glycerol sebacate)-modified polylactic acid scaffolds with improved hydrophilicity, mechanical strength and bioactivity for bone tissue regeneration
Hengsong Shiab,
Qi Gan*ab,
Xiaowei Liuc,
Yifan Maab,
Jun Huc,
Yuan Yuan*ab and
Changsheng Liu*ab
aThe State Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai 200237, PR China. E-mail: ganqi@ecust.edu.cn; yyuan@ecust.edu.cn; liucs@ecust.edu.cn
bKey Laboratory for Ultrafine Materials of Ministry of Education, East China University of Science and Technology, Shanghai 200237, PR China
cState Key Laboratory of Chemical Engineering, Department of Chemistry, East China University of Science and Technology, Shanghai, China
First published on 14th September 2015
Abstract
Polylactic acid (PLA) has been extensively researched in biomedical engineering applications due to its superior mechanical strength and biocompatibility in vivo. But the inherent brittleness, slow degradability and inferior hydrophilicity greatly hamper its successful application. Here, a biodegradable crosslinked elastomer poly(glycerol sebacate) (PGS) was adapted to modify PLA scaffold for bone tissue engineering in this study. A highly interconnected and large porous, three-dimensional (3D) PLA-based scaffold was prepared by a NaCl particulate-leaching method and the PGS prepolymer (pre-PGS) was introduced either by pre-molding binary blend (B.B) or by surface coating (S.C) of a homogeneous PGS onto PLA-based scaffolds with and without oxygen plasma pretreatment (O.P and D.C). After curing at 130 °C, the resulting PLA/PGS scaffolds all exhibited well interconnected open-cell structures. The incorporation of PGS to PLA both by B.B and S.C could effectively improve the hydrophilicity, degradation, toughness and ductility, and the best efficacy was observed for the S.C with the oxygen plasma pretreatment. Specifically, at the ratio of PLA/PGS 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 7:3, the fracture strain of the PLA/PGS scaffolds by O.P were improved from 8% (pure PLA) to 13% and 24%, respectively. Further studies indicated that enhanced hydrophilicity and increased surface roughness were the main contributors to the above positive effect of oxygen-based plasma treatment. Additionally, these hybrid PLA/PGS scaffolds exhibited good mineralization, high cell biocompatibility, and enhanced cell adhesion and osteogenic differentiation for bone mesenchymal stem cells (BMSCs), especially for scaffolds by S.C. The present results suggest that the surface coating of PGS with oxygen-based plasma pretreatment is an effective strategy to modify the properties of PLA and the hybrid PLA/PGS scaffold represents a promising candidate in the formulation of bone tissue regeneration.
:1 and 7:3, the fracture strain of the PLA/PGS scaffolds by O.P were improved from 8% (pure PLA) to 13% and 24%, respectively. Further studies indicated that enhanced hydrophilicity and increased surface roughness were the main contributors to the above positive effect of oxygen-based plasma treatment. Additionally, these hybrid PLA/PGS scaffolds exhibited good mineralization, high cell biocompatibility, and enhanced cell adhesion and osteogenic differentiation for bone mesenchymal stem cells (BMSCs), especially for scaffolds by S.C. The present results suggest that the surface coating of PGS with oxygen-based plasma pretreatment is an effective strategy to modify the properties of PLA and the hybrid PLA/PGS scaffold represents a promising candidate in the formulation of bone tissue regeneration.
Introduction
In recent decades, an increasing number of bone defects caused by an aging population, bone necrosis, bone cancer and other diseases have greatly driven the development of biomaterials for bone tissue regeneration.1,2 In general, bone repair scaffolds should be biocompatible, biodegradable, and osteoconductive and should have appropriate biomechanical properties during the regeneration of the tissue.3,4 With these respects, a series of synthetic polymers, including polyhydroxybutyrate (PHB), polycaprolactone (PCL), and polyurethane (PU), are widely developed in bone repair engineering field.5,6 Among them, polylactic acid (PLA), owing to its high mechanical properties, easy processability and biocompatibility in vitro and in vivo, has been widely investigated and applied.7–9 However, the inherent brittleness, low hydrophilicity and biodegradation rates have limited its biological application.9–11 Incorporating other polymers or nano-fillers, such as bioglass and hydroxyapatite, is considered as a promising method to improve the properties of original PLA scaffolds.3,8,12,13Poly(glycerol sebacate) (PGS), a covalently crosslinked elastomer based on the polymerization of glycerol and sebacic acid, due to super elasticity, in vivo linear degradation and relatively appropriate hydrophilicity, has been widely explored in the field of soft tissue replacement and engineering,14,15 such as nerve guide, vascular graft, lung and heart valve.16 More importantly, PGS can be prepared as a pre-polymer by polycondensation of equimolar mixtures of glycerol and sebacic acid, followed by a heat treatment (120 °C) to form crosslinked PGS elastomers, allowing for their easy processability for fabrication or modification of 3D scaffolds.15,17 Inspired by these excellent properties associated with PGS, we endeavor to investigate the possibility of utilizing the crosslinked PGS to improve the physicochemical properties and biological functions of PLA and widen its applications in bone tissue regeneration.18 To the best of our knowledge, only several reports have focused on the PLA and PGS composites to date.9,19 But their studies mostly used PLA as minor second polymer to improve the properties of PGS matrix phase and thus widened its application range in soft tissue repair.11,20,21 And due to the poor mechanical strength, these PLA-modified PGS-matrix composite has never been used as bone tissue engineering.
Herein, we endeavoured to use the super elasticity of PGS to improve the toughness of the conventional PLA for bone tissue engineering. To meet the basic requirement of mechanical strength of bone tissue engineering, high molecular weight PLA (Mn = 1.2 × 105 g mol−1) with a melting temperature (Tm) of approximately 166 °C was applied as matrix and the ratio of PLA to PGS was fixed at 9:1 and 7:3. PGS pre-polymer was synthesized via polycondensation and further applied to modify PLA scaffold for bone tissue regeneration. PGS pre-polymer modification has been achieved via three simple approaches (Fig. 1): (i) pre-molding binary blend (B.B) and (ii) post-molding surface coating without and with oxygen plasma pretreatment (D.C and O.P). The effects of mixture ratio and preparation methods on the morphology, mechanical strength as well as the interface of the resulting scaffolds were investigated. Furthermore, with bone mesenchymal stem cells (BMSCs) as an in vitro cell model, we further evaluated cell attachment and cytotoxicity on these scaffolds.22 It is anticipated that our PGS modification could provide valuable strategy to improve the properties of PLA for bone tissue regeneration.
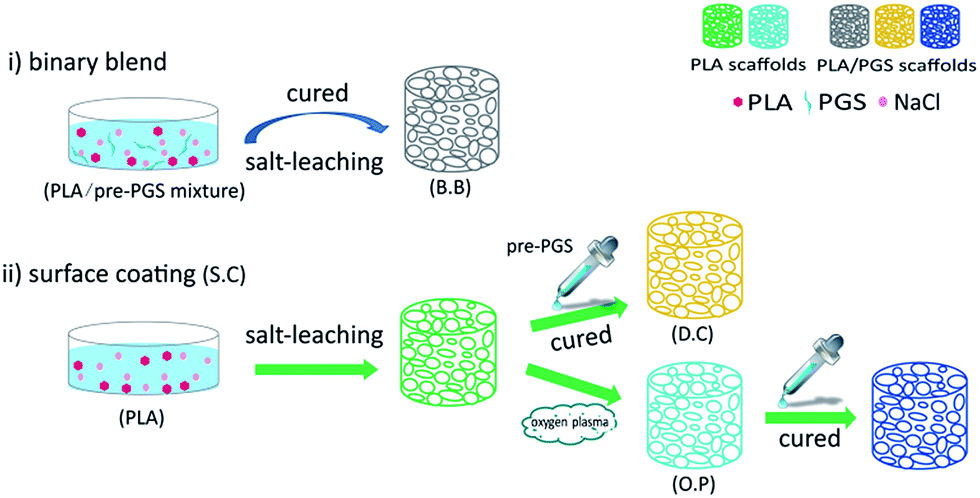 | ||
| Fig. 1 Schematic diagram of three different approaches for PLA/PGS scaffolds. (i) Pre-molding binary blend (B.B): PLA and pre-PGS were mixed directly and PLA/PGS porous scaffolds prepared by a NaCl particulate-leaching technique. (ii) Surface coating (S.C): PLA-based porous scaffolds were prepared first, and then coated with specific proportion of PGS directly (D.C) and PLA-based porous scaffolds surface treated with oxygen plasma, finally coated with PGS (O.P). All resulting PLA/pre-PGS scaffolds were cured at 130 °C in a vacuum oven for 48 h. | ||
Experimental
Materials
PLA (4032D) (Tg = 57.8 °C, Tm = 166 °C, Mn = 1.2 × 105 g mol−1) was supplied by NaturePlast (America). Sebacic acid (99.0% purity, Aladdin), glycerol (≥99.0% purity, Sigma-Aldrich), Ar (99.99% purity, Air Liquid) were used as received, ethanol (99.7% purity), dichloromethane (99.5% purity), chloroform-D (99.8% purity) and tris-(hydroxymethyl)-aminomethane solution (pH = 7.4) (Tris–HCl, 99.0% purity) were all bought from Sinopharm Chemical Reagent Co., Ltd, (Shanghai, China). All other reagents used in cell culture were from Gibco (Grand Island, NY).Synthesis of poly(glycerol sebacate) pre-polymer (pre-PGS)
PGS pre-polymer was prepared from the polycondensation reactions of equimolar glycerol and sebacic acid according to previously published methods. Briefly, equimolar amounts of glycerol and sebacic acid (1:1) were melted at 130 °C and stirred in a 100 mL three necked round bottom reactor under Argon atmosphere for 36 h. Then the reaction was conducted under vacuum and pressure reduced from 1 Torr to 50 mTorr over 2 h. Finally, further polymerization was continued for 6 h at 160 °C. The obtained viscous mixture was dissolved in ethanol and purified by ultrapure water, subsequently freeze-dried in the lyophilizer and obtained the resulting PGS pre-polymer (pre-PGS).
Preparation of PLA and PLA/PGS (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1, 7:3) porous scaffolds
:1, 7:3) porous scaffolds
PLA and PLA/PGS (PLG) porous scaffolds were fabricated by a NaCl particulate-leaching technique. The thickness, porosity and pore size could be easily controlled by the total weight, NaCl particle proportion and size, respectively. PLA was dried in a vacuum chamber at 80 °C in advance. As mentioned above, the PLA offered the mechanical strength. When the content of PGS was higher than the PLA, scaffolds exhibited lower strength and other mechanical properties, which was not suitable for bone tissue regeneration. Therefore, PLA/PGS at the ratio of 9:1 and 7:3 were selected for our following research. Three different approaches were adopted for scaffolds composite as shown in Fig. 1. Binary blend (B.B): pure PLA, PLA/pre-PGS (9:1) and PLA/pre-PGS (7:3) (abbreviated as PLG9 and PLG7) were obtained and dissolved in 30 mL dichloromethane. All solutions were prepared separately in closed glass containers and stirred for 2 h at room temperature. NaCl particles (0.2–0.4 mm), 80 wt% were added to each solution. The mixtures were cast into round molds (15 × 15 × 5 mm3) and air dried overnight to allow solvent evaporation. After that, the mixtures and the pure PLA were all cured at 130 °C for 48 h and were subsequently immersed in ultrapure water at 60 °C for 48 h to leach NaCl granules out. Surface coating (S.C) was also employed to composite PLA with PGS. PLA porous scaffolds were first prepared, and then coated with the corresponding proportion of PGS directly (namely D.C) or with oxygen plasma pretreatment (CTP-2000K, power = 40 W, time = 90 s) (namely O.P). After that, the pre-PGS/PLA scaffolds were repeated the above curing and salting-out process. Raw material compositions of scaffolds were given in Table 1.
| Samples | PLA (wt%) | PGS (wt%) | Salt (0.2–0.3 mm) |
|---|---|---|---|
| PLA | 20 | 0 | 80% |
| PLG9 | 18 | 2 | 80% |
| PLG7 | 14 | 6 | 80% |
Physiochemical characterization
To investigate the surface hydrophilicity of PLA before and after oxygen plasma treatment, static contact angle measurements using ultrapure water were conducted.
:1, 7:3) blends were dissolved in dichloromethane and cast on a non-stick baking tray, respectively. The cast samples were dried in the fume hood overnight, and cured under vacuum at 130 °C for 24 h for preliminary crosslinking, these semi-crosslinked samples were removed and pressed into smooth sheets. Finally cured under vacuum at 130 °C for another 24 h. Contact angles of static water in air were measured using the sessile drop method on a contact angle measure instrument Phoenix 300 (Korea). Droplets of ultrapure water (4 μL) were extruded to the surface of the sheets. The contact angle was obtained by measuring the angle between the sample surface and a tangent to the water drop surface. Images of the droplets were captured with a CCD camera, and the contact angle values were analyzed by an image acquisition system.Impact strength test was carried out on Universal Pendulum Impact Tester (F2056, Jiangsu, China) according to GB/T 1843-2008 standard.
To prove the applicability of scaffolds in bone tissue regeneration, not only the mechanical test, but the degradation of materials in vitro, mineralization and cell differentiation were characterized.
In vitro mineralization and degradation
Simulated body fluid (SBF) was prepared according to reports published previously, and that required sterilization before using. PLA and PLG porous scaffolds were soaked in SBF and incubated in a controlled environment (thermostated at 37 °C) for 3 days.The cured PLA and PLG9 and PLG7 porous scaffolds were made into disks with a diameter of 15 mm and a thickness of 5 mm (15 × 15 × 5 mm3). All dried specimens were immersed in a tris-(hydroxymethyl)-aminomethane (Tris–HCl) solution (pH = 7.4) and thermostated at 37 °C, with a mass/volume ratio of 1 g/20 mL. The Tris–HCl solution was renewed every two days in the first 20 days, it was every 10 days after that. And the pH value of the degradation medium was monitored after each solution change. After predetermined intervals of time, the samples were removed, washed with ultrapure water and freeze-drying till constant weight. The weight loss of these scaffolds with the time of degradation was calculated with the equation followed:
 | (1) |
In vitro cell studies
Statistical analysis
All numerical data were expressed as the mean ± standard deviation. Statistical analysis was performed with one-way analysis of variance (ANOVA). A value of p < 0.05 was considered as statistical significance.Results and discussion
Synthesis and characterizations of pre-PGS and PGS/PLA scaffolds
A hyperbranched PGS pre-polymer was synthesized by polycondensation of equimolar glycerol and sebacic acid (Fig. 2(A)). The pre-PGS with small molecular weight was purified by dialysis membrane (MWCO: 3000 Da) against water, and subsequently lyophilized to get pre-polymer as yellowish viscous liquid (pre-PGS).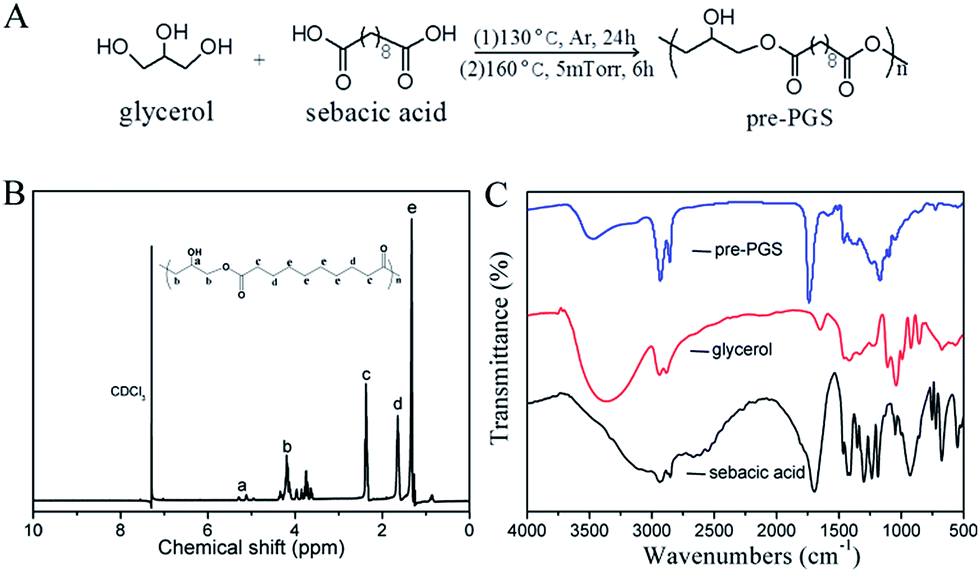 | ||
| Fig. 2 Synthesis and characterizations of poly(glycerol sebacate). (A) PGS pre-polymer (pre-PGS) was synthesized through polycondensation of equimolar glycerol and sebacic acid. (B) 1H-NMR spectrum of pre-PGS and (C) FTIR spectra of the pre-PGS, glycerol and sebacic acid. | ||
In order to retain a high ratio of free hydroxyl groups attached to the pre-PGS backbone for subsequent interaction with PLA, the polycondensation of pre-PGS was terminated prior to its anticipated gel point. In this experiment, the pre-PGS with a low molecular weight Mn of 4000 g mol−1 and a polydispersity index of 3 were determined by GPC. The incorporation of the sebacoyl moiety in PGS molecular chain was confirmed by 1H-NMR (Fig. 2(B)) by the appearance of the peaks at δ 1.33, 1.72 and 2.25 ppm, respectively. Additional peaks at δ 3.5–5.5 ppm identified in the pre-PGS spectrum corresponded to the glyceryl moiety in PGS molecular chain.23 The typical absorption peaks of pre-PGS, glycerol and sebacic acid were characterized by FTIR spectra. As shown in Fig. 2(C), typical absorption bands of hydroxyl (3200–3500 cm−1) and ester (1600–1800 cm−1) groups were observed in the backbone of PGS. Sebacic acid presented large absorption peaks at 1730 cm−1 and 1400 cm−1 (asymmetric and symmetric stretching peaks of the carboxylate salt groups). While the intensity of these peaks significantly declined, and the ester linkage (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) attached to the pre-PGS backbone shifted to 1750 cm−1 after polycondensation. Overall, pre-PGS with a relatively low molecular weight was successfully synthesized.
O stretching) attached to the pre-PGS backbone shifted to 1750 cm−1 after polycondensation. Overall, pre-PGS with a relatively low molecular weight was successfully synthesized.
NaCl particulate-leaching technique was commonly adapted to obtain sponge-like scaffolds with macroporous structure. The surface morphologies of PLA, O.P treated PLA and PLG porous scaffolds after cured were examined by SEM, as presented in Fig. 3(A). PLA and PLG porous scaffolds all exhibited well open-cell structure and high connectivity.9 Macropores left behind by NaCl crystals after leaching were evident on PLA and PLG scaffolds, which are considered to be beneficial for cellular adhesion and nutrient transmission. Additionally, the differences of the porosity, pore size and microstructure probably resulted from the size and shape of NaCl crystals.
 | ||
| Fig. 3 Microstructural analysis of PLA and PLG scaffolds. (A) SEM micrographs of pristine PLA, O.P treated PLA, PLG9 and PLG7 scaffolds prepared via B.B, D.C and O.P, presenting highly interconnected open-cell structure. (B) Phase interfacial morphology and distribution of PLA, PLA-O.P and PLG9 as well as PLG7 scaffolds by B.B, D.C and O.P (scale bar: A = 500 μm; B = 50 μm). | ||
As depicted in Fig. 3(A), the cell-walls of pure PLA, O.P treated PLA and PLG scaffolds by B.B seemed thinner than PLG scaffolds prepared via D.C and O.P. Moreover, the cell-walls of PLG7 scaffolds looked thicker than PLG9 scaffolds, and the thickness increased with increasing of pre-PGS content. The cause for this effect might be explained that the pre-PGS was post-coated onto the porous PLA-based scaffolds by infiltration technique, higher concentration of pre-PGS rendered PLG7 scaffolds achieved higher average pore thickness. For the scaffolds prepared via D.C and O.P, plenty of white substances can be found around the edge of these pores, which presumably attributed to the distribution of the phase of PGS. White spots on D.C and O.P prepared scaffolds were more obvious than B.B. The phenomenon was more notable with higher pre-PGS content (PLG7).
In this work, PLA and PLA/PLG scaffolds were subjected to harsh curing condition (48 h at 130 °C).24 We first studied the effect of the curing process on the degradation of PLA. GPC of PLA before and after the curing process (130 °C for 48 h) were tested. According to the GPC results, the molecular weight of PLA after thermal curing decreased slightly to ∼1.05 × 105 g mol−1. The extent of PLA degradation is not very obvious during curing process and has little effect on the results of the experiment later. That may be ascribed to the high molecular weight of PLA (Mn = 1.2 × 105 g mol−1) with a melting temperature (Tm) of approximately 166 °C, which is much higher than the curing temperature (130 °C). Meanwhile, from the SEM micrographs, PGS and PLA were well distributed throughout the scaffold matrix, and both components can coexist in a hybrid system. The results proved that there had no significant pore collapse due to the presence of PLA with high Tm. Furthermore, the surface appeared to be smoother with increasing pre-PGS content.
The morphology and distribution of PLG scaffold interfaces were further observed in Fig. 3(B), with pure PLA and O.P treated PLA scaffolds as control groups. The surface of PLA scaffold was rather rough and intricate at high magnification, more coarse and etched traces can be observed on PLA-O.P. While PGS-modified PLA scaffolds showed different morphologies at the same magnification. A lot of cracks, even large fracture surface can be found on B.B prepared PLG scaffolds, and PGS haphazard embedded in PLA phase. The analysis indicated that PGS and PLA irregularly distributed throughout the scaffolds. PLG scaffolds prepared via D.C and O.P were post-coating pre-PGS on the surface of PLA-based porous scaffolds. However, a much more homogeneous coating of PGS on the O.P-treated PLA scaffolds were observed in Fig. 3(B). Scaffold prepared via O.P exhibited a smoother and more uniform surface. For the scaffold by D.C, an irregularly distributed PGS phase distributed on the PLA surface and the interface of PLA–PGS could be clearly identified as a result of phase separation occurring in the course of curing. The SEM analysis, especially the investigation of the interface illustrated that the addition of PGS onto PLA-based scaffolds had an obvious effect on the morphology of PLG scaffolds, and the way of PGS be introduced similarly had a decisive role on the interface between these two polymers. Additionally, the results also indicated that the oxygen plasma treatment could induce pre-PGS evenly coated on the surface of PLA-based scaffold.
In order to examine whether a chemical reaction occurred between PLA and PGS, FT-IR analysis was performed and the results are shown in Fig. 4. The typical absorption peak at approximately 3500 cm−1 (H-bonded hydroxyl group) had been observed, and the intensity of the peak increased with the increasing weight percentage of PGS. Previous studies of PLA and PGS analysis with the help of FT-IR spectrum revealed the stretch vibration peaks at 3000 cm−1 (due to PLA), 2926 cm−1, and 2852 cm−1 (due to both PLA and PGS), representing the alkyl groups. The intense absorption peaks at 1730 cm−1 and 1160 cm−1 indicate the double bond (CO) and C–O (due to both PGS and PLA). The results demonstrated that a simple physical mixing process occurred during the curing process.
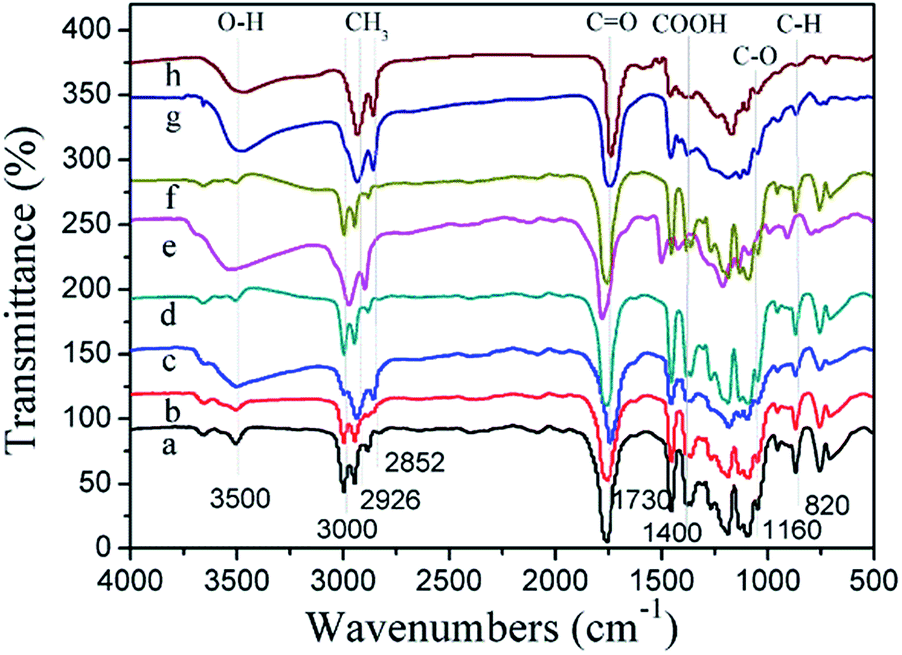 | ||
| Fig. 4 FT-IR spectra of PLA (a), PLG9 scaffolds by B.B (b), PLG7 scaffolds prepared via B.B (c), D.C prepared PLG9 scaffolds (d), PLG7 scaffolds by D.C (e), PLG9 scaffolds by O.P (f), PLG7 scaffolds prepared via O.P (g) and pure PGS (h) cured blends at 130 °C. It indicates that there are no functional groups disappeared and new functional groups generated, which shows a simple physical mixing process occurred during the curing process. | ||
Hydrophilicity of the prepared scaffolds
Pure PLA has poor hydrophilicity as reported previously,25 which greatly hampers its targeted applications for various biomedical engineering fields. PGS was traditionally considered as a hydrophilic material owing to hydroxyl groups attached to the structure backbone.As shown in Fig. 5, the hydrophobic surface of the pristine PLA showed a contact angle of 101° ± 3.3°, and 69° ± 3.6° for PGS. As anticipated, the hydrophilic property of pristine PLA significantly improved with the introduction of PGS. It showed that the contact angles of PLG9 and PLG7 prepared via B.B declined by 12% and 19% points, respectively. PLG9 and PLG7 scaffolds by D.C showed contact angles of 70° ± 2.5° and 73° ± 2.3°. The contact angles reduced to 71° ± 2.8° and 69° ± 1.7° as a consequence of the oxygen-based plasma surface modification of O.P prepared PLG9 and PLG7 scaffolds. Furthermore, it can be observed that the contact angles of PLG9 and PLG7 scaffolds by D.C and O.P were smaller than that the corresponding B.B. No significant discrepancy between D.C and O.P prepared PLG scaffolds in the contact angle results due to the PGS layer on PLA surface. These results indicated an increase in the hydrophilic ability of PLG scaffolds with the addition of PGS. Moreover, PLG scaffolds fabricated by surface coating exhibited better hydrophilicity compared to that obtained by B.B process.
 | ||
| Fig. 5 Hydrophilic analysis of PLA, PGS and PLG scaffolds. (A) Water drop profiles of the pristine PLA, PGS and PLG scaffold surfaces prepared via B.B, D.C and O.P and (B) contact angle measurements for these scaffold surfaces. | ||
Mechanical properties of the prepared scaffolds
Mechanical durability plays an important role in the tissue engineering applications of bone defects, especially for hard tissue repair scaffolds.26 Despite of the splendid mechanical properties and biocompatibility in vitro, pure porous PLA scaffolds still can't meet the need of load-bearing bone repair fields due to its brittleness. Table 2 presents the results of fracture stress (MPa) and fracture strain (%) for PLA, O.P treated PLA and PLG scaffolds by B.B and S.C. Representative linear fracture stress–strain curves for these scaffolds are depicted in Fig. 6, a and b curves represented the pure PLA and PLA-O.P scaffolds, c and d curves were scaffolds treated with oxygen plasma, d and g curves present scaffolds by D.C, curves by B.B were e and h, respectively. Additionally, c, d and e curves stand for scaffolds of PLG9, f, g and h were curves for PLG7 scaffolds. The results demonstrated the elastomeric nature of PGS can optimize the toughness and ductility of PLA scaffolds.| Samples | B.B | D.C | O.P | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Fracture stress (MPa) | Fracture strain (%) | Impact strength (kJ m−3) | Fracture stress (MPa) | Fracture strain (%) | Impact strength (kJ m−3) | Fracture stress (MPa) | Fracture strain (%) | Impact strength (kJ m−3) | |
| PLA | 1.72 ± 0.3 | 8 ± 2.2 | 5.2 ± 0.8 | — | — | — | — | — | — |
| PLA-O.P | 1.57 ± 0.2 | 8.5 ± 2 | 5.3 ± 0.6 | — | — | — | — | — | — |
| PLG9 | 1.1 ± 0.24 | 7 ± 1.2 | 4.8 ± 0.6 | 1.52 ± 0.31 | 13.3 ± 1.42 | 5.4 ± 1.1 | 1.58 ± 0.27 | 13 ± 1.31 | 7.7 ± 0.5 |
| PLG7 | 0.9 ± 0.2 | 7 ± 1.6 | 3.9 ± 1.2 | 1.41 ± 0.12 | 20 ± 1.54 | 4.7 ± 0.6 | 1.6 ± 0.23 | 24 ± 1.8 | 6.5 ± 1.2 |
 | ||
| Fig. 6 Compressive stress–strain curves of PLA (a), O.P treated PLA (b), PLG scaffolds with various PGS ratios and preparation methods: (c–e): PLG9; (f–h): PLG7; (c and f) prepared via O.P; (d and g) prepared via D.C; (e and h) prepared via B.B. | ||
As presented in Table 2, it is evident that the pure PLA scaffold obtained the higher fracture stress (1.72 ± 0.3 MPa) and relatively lower fracture strain (8 ± 2.2%). PLA treated by oxygen plasma presented relatively higher elongation at break (8.5 ± 2%) and lower fracture stress (1.57 ± 0.2 MPa) than the pristine PLA. Curiously, PLG9 and PLG7 scaffolds by B.B exhibited poor fracture stress (approximately 1 MPa) and fracture strain (7%). As expected, the strain at break of PLA scaffolds increased with an increase in PGS content. However, no significant improvement was observed for B.B prepared PLG scaffolds. The fracture stress of PLG scaffolds by S.C slightly decreased with the introduction of PGS compared with the pristine PLA scaffold, while the strain at break largely improved. For example, the fracture stress of PLG9 and PLG7 scaffolds prepared via D.C were 1.52 ± 0.31 MPa and 1.41 ± 0.12 MPa, the fracture strain were 13.3 ± 1.42% and 20 ± 1.54%, respectively. Specifically, scaffolds treated with oxygen plasma (O.P) exhibited higher compressive strength (1.58 ± 0.27 MPa and 1.6 ± 0.23 MPa) and strain (13 ± 1.31% and 24 ± 1.8%). Which was almost 3-fold observed in strain at break of PLG7 scaffold compared to PLA scaffold. It is interesting to note that, O.P-treated scaffolds obtained a higher mechanical strength and ductility than D.C under the same conditions, the only difference existed in the composition of PLA and PGS is that the O.P prepared scaffold had been pre-modified by oxygen-based plasma treatment, which significantly reinforced the mechanical properties. The results indicated that the oxygen plasma treatment had obvious effects on the mechanical strength of PLG scaffold. A relatively ductile and tough PLA-based scaffold combined with super elasticity of biodegradable PGS was producible.
The elastomeric nature of PGS may also affect the impact strength values of the PLA scaffolds. Table 2 also lists the values of the impact strengths of PLG9 and PLG7 scaffolds prepared via B.B, D.C, O.P and pure PLA porous scaffold. These results suggested a general tendency that PLG scaffolds by S.C with the oxygen plasma treatment obtained the highest impact strength (7.7 ± 0.5 kJ m−3) compared with PLA scaffold among these three approaches, whereas a slight decrease can be observed with PGS concentration increased.
The FT-IR results also demonstrated the changes of mechanical properties mainly due to the introduction of PGS rather than a chemical reaction occurred during the composition of PLA and PGS.
The physical mechanism of PGS affected the mechanical properties of PLA can be illustrated by DSC results.27 Table 3 lists the thermal data and Fig. 7 presents the DSC crystalline curves of PLA, PLG9 and PLG7 cured blends. As depicted in Fig. 7, the glass transition temperature (Tg), crystallization temperature (Tc) and melting temperature (Tm) of pristine PLA were very high, reflecting that the moving capability of molecular chain segments of the PLA matrixes was weak. With the increasing ratio of PGS, Tg, Tc and Tm decreased monotonically, which indicated that the moving capability of PLG molecular chain segments was strengthened. This was presumably ascribed to the introduction of PGS, which partly penetrated into PLA molecular chain. PGS performed as a softening agent in the PLG blend, effectively weakened the intramolecular interaction of PLA. Overall, the introduction of PGS strengthened the moving capability of PLA molecular chain and rendered better ductility and toughness.
| PLG (weight ratio/g g−1) | Tg (°C) | Tc (°C) | Tm (°C) |
|---|---|---|---|
| PLA | 59.75 | 126.68 | 166.39 |
| PLG9 | 55.33 | 109.18 | 166.59 |
| PLG7 | 53.42 | 107.58 | 165.97 |
 | ||
| Fig. 7 DSC crystalline curves of PLA, PLA/PGS (9:1, 7:3) cured blends. | ||
XPS analysis and surface morphology after oxygen plasma treatment
Above results indicated that the oxygen plasma treatment favored for the improvement of the hydrophilicity and mechanical strength of the PLG/PLA scaffolds. In order to explore the underlying mechanism involved, in the following experiment, the effect of oxygen-based plasma treatment was researched by XPS, water contact angle and AFM.26,28,29The chemical compositions and structure of untreated and plasma-treated PLA surfaces identified by XPS analysis are displayed in Fig. 8(A). Fig. 8(A)-a showed the XPS full spectra from untreated PLA and PLA surface treated with oxygen plasma, and exhibits two peaks located at binding energies of 283 and 531 eV, corresponding to C1s and O1s, respectively. Fig. 8(A)-b shows the C1s spectra of untreated PLA, it was separated to three C1s peak components of approximately the same composition, corresponding to the three types of carbon atoms presented in PLA. The first peak at 285.0 eV corresponded to C–C/C–H. The second peak appeared at 286.6 eV represented C–O–C groups. The last one at a binding energy of 289 eV, corresponding to O–CO groups, in which the carbon atom was double bonded to oxygen atom. Almost the same properties can be found on the C1s peak of PLA surface treated with oxygen plasma as shown in Fig. 8(A)-d. Remarkable changes can be found compared C1s (b) with (d) before and after oxygen plasma treatment. The ether peak (C–O–C) and ester peak (O–CO) increased by 20.1% and 40.2% with the simultaneous 26.9% decrease of C–C/C–H peak. Table 4 gives the percentages and binding energies of atomic compositions for PLA surface before and after oxygen plasma treatment. The results calculated from the XPS results (Table 4) showed that the oxygen-based plasma treatment was able to improve the ratio of oxygen-containing functional groups on the modified surface of PLA (e.g. C–O, O–CO) and the O/C atomic ratio of PLA treated with oxygen plasma (86.4%) was 35.7% higher than untreated (50.7%). The results suggested that the enrichment of oxygen-containing functional groups was understood as a consequence of the oxygen plasma treatment.
 | ||
| Fig. 8 Surface analysis of PLA before and after oxygen plasma treatment. (A) XPS survey scan spectra: (a) XPS full spectra from untreated PLA and PLA surface treated with oxygen plasma; (b and c) XPS full spectra from untreated PLA, (b) C1s XPS spectra and (c) O1s XPS spectra; (d and e) XPS full spectra from treated PLA, (d) C1s XPS spectra and (e) O1s XPS spectra; (B) water drop profiles (left), contact angles of original PLA and plasma-treated PLA films (right). | ||
| Samples | XPS elemental ratio (%) | XPS C1s envelope ratios (%) | XPS O1s envelope ratios (%) | ||||
|---|---|---|---|---|---|---|---|
| C | O | C–C/C–H | C–O–C | O–CO |
OC |
O–C | |
| Binding energy (eV) | Binding energy (eV) | ||||||
| 285.0 | 286.6 | 289 | 532.39 | 533.85 | |||
| Untreated | 66.37 | 33.63 | 50.7 | 30.72 | 18.58 | 60.02 | 39.98 |
| Treated | 53.65 | 46.35 | 37.05 | 36.9 | 26.05 | 44.57 | 55.43 |
Following the oxygen-based plasma surface modification, the contact angles of PLA surface before and after oxygen plasma treatment were conducted. As seen in Fig. 8(B), contact angles decreased from 101° ± 3.4° to 32° ± 2°, which comprised a hysteresis of 5°. Thus the results indicated the oxygen plasma treatment could further render the PLA surface to become more hydrophilic, this analysis was consistent with the results of XPS.
Not only that, oxygen plasma treatment could increase the roughness of PLA surface due to the etching effect produced by the bombardment of high energy particles (e.g. electrons, radicals, ions, neutrals atoms, etc.) reported by many researchers.30,31 Similar effects of PLA surface treated with oxygen-based plasma had also been confirmed by AFM analysis. Fig. 9 shows typical AFM topographies of PLA films before and after oxygen plasma treatment. The original PLA surface (A) presented a remarkably smooth surface with a root mean square roughness (RMS) of 20.965 nm, which was almost seventh of plasma-treated PLA surface (B). The RMS roughness was 152.82 nm. As depicted in Fig. 9(B), PLA surface presented many needle-like structures after oxygen plasma treatment.
 | ||
| Fig. 9 Representative AFM images of untreated and plasma-treated PLA films. (A) Original PLA and (B) plasma-treated PLA surfaces. | ||
It can be observed from the analysis of XPS and water contact angle, together with AFM, that the oxygen plasma treatment effectively modified the microtopography of PLA surface. The hydrophilicity increased and the surface turned rougher. Furthermore, such physical changes may allow for a homogeneous coating of PGS tightly covered on the PLA surface, and then the strengthened PLG scaffolds were obtained.
In vitro mineralization and biodegradation
Biomineralization ability is a key factor to promote the osteocompatibility and bone-binding ability of biomaterials for bone regeneration. The mineralization ability of the scaffolds is generally evaluated by their chemical composition changes and specific surface structure after biological soaking treatment. Herein, a 7 day mineralization of PLA and PLG scaffolds was investigated in vitro and the corresponding morphologies are given in Fig. 10(A). | ||
| Fig. 10 Mineralization analysis of PLA and PLG scaffolds. (A): Representative SEM images showing the mineralization morphology of PLA (a), plasma treated PLA (b), PLG9 scaffolds by B.B (c), PLG7 scaffolds prepared via B.B (d), D.C prepared PLG9 scaffolds (e), PLG7 scaffolds by D.C (f), PLG9 scaffolds by O.P (g), PLG7 scaffolds prepared via O.P (h); XRD patterns of scaffolds prepared via D.C before (B) and after (C) mineralization (scale bar: 10 μm). | ||
There is no significant difference in mineralization of these materials. Bulk and nanorod-like minerals sparsely distributed on the surface of scaffolds. Massive mineralized deposits are observed in Fig. 10(A)-c. In order to identify these deposits, XRD measurement was conducted. The relevant patterns are presented in Fig. 10(B) and (C). In this study, all scaffolds including pure PLA and PLA-O.P were soaked in SBF for mineralization, while another untreated PLG7 scaffold by O.P was set as a control group. XRD patterns are shown in Fig. 10(C). PLA was semi-crystalline, the main peak located at 16.8°, other weak peaks appeared at 19.1° and 22.5° are seen in Fig. 10(C).32 PGS was amorphous, which signal peaks were too weak to be distinguished. The XRD pattern of scaffolds exhibited several particular peaks after mineralization. A remarkable peak at 2θ = 31.8° was observed.6 Based on previous literatures, this location was the peak position of hydroxyapatite (HA). However, no significant signs of mineralization could be found in the control group. The results were consistent with the SEM analysis (left) and demonstrated the capability of inducing bone-like apatite formation of PLG scaffolds.
In vitro degradation of PLA and PLG scaffolds fabricated by B.B and S.C was carried out to observe the pH, mass loss and morphology changes. The results are depicted in Fig. 11. All the samples were immersed in Tris–HCl medium for a maximum of 50 days. The changes of pH were recorded in the first 25 days, pH change profiles at different time points are illustrated in Fig. 11(A). It is apparent that the pH significantly decreased with the addition of PGS compared with the pure PLA scaffolds. The pH values of pristine PLA scaffold barely changed and stayed at 7.4. Previous in vivo degradation studies had demonstrated that PLA poses an extremely slower degradation rate compared with PGS, and it took at least 2 years to degrade completely in vivo.9,33 Simultaneously, PLG scaffolds fabricated by S.C (D.C and O.P) dropped faster than B.B and the pH was lower than 7.2 after 25 days. Overall, a decreasing tendency can be observed in PLG scaffolds might due to the release of the acidic product from PGS and thus accelerated the degradation rate of PLG scaffolds.
 | ||
| Fig. 11 In vitro degradation of PLA and PLG9 and PLG7 scaffolds fabricated via B.B, D.C and O.P. (A) pH, (B) mass loss, (C) SEM and (D) high magnification images of 50 days of prepared scaffolds with degradation time (scale bar in C: 20 μm, scale bar in D: 10 μm). | ||
Mass loss profiles of porous scaffolds during degradation are displayed in Fig. 11(B). The mass loss profiles of all scaffolds were nearly linear with the increasing degradation time. Similarly, the weight loss rate of PLG scaffolds obviously increased with the introduction of PGS compared with the pure PLA scaffolds, which degraded only approximately 2% after 50 days in Tris–HCl.
Meanwhile, PLG scaffolds by S.C degraded significantly faster than B.B might be attributed to the large surface of PGS coated on the PLA substrate. And scaffolds prepared via O.P had relatively slower degradation rates than D.C, which can be explained that a homogeneous and continuous coating of PGS distributed on the surface of PLA scaffolds may not only allow for self-crosslinking of PGS via their strong intramolecular interaction, but also result in an improvement of the composition of PLA and PGS after oxygen plasma treatment. Such structure finally slowed the degradation rates of scaffolds by O.P.
Fig. 11(C) shows the morphology of degraded samples at different degradation time points. All PLG scaffolds presented a more drastic erosion profile than the pure PLA scaffolds, which might be attributed to the release of the small molecules from PGS and leading an acceleration of the degradation of PLG scaffolds. And the scaffolds by S.C presented more obvious degradation morphology than B.B. It also can be observed from this figure that scaffolds prepared via D.C degraded faster than O.P with high magnification of 50 days (Fig. 11(D)). Furthermore, the analysis of the surface morphology demonstrated the signs of surface degradation, characterised by craters and pits, erosion pits on the surface of samples increased gradually with the increased degradation time, which might due to the linear degradation of PGS in vivo through a surface erosion mechanism as reported previously.34
Cytocompatibility and cell adhesion of the prepared scaffolds
The cytocompatibility of the PLG scaffolds was measured with the MTT assay, a widely used method to measure the mitochondria activity to quantify the cell growth/death. As shown in Fig. 12, the cytocompatibility of PLA, PLA-O.P and six PLG groups all show no significant difference to the control group (p > 0.05), with the similar trend after 1 day and 3 days exposure. These cytocompatibility results demonstrated that our composite scaffolds exhibited no toxicity to the BMSCs in vitro, which can be attributed to the high biocompatibility of PLG materials. | ||
| Fig. 12 Cytocompatibility of PLA, PLA-O.P and composite PLG scaffolds cultured with BMSCs for 1 day and 3 days (n = 4, *p < 0.05). | ||
In general, cell adhesion is the first step of the cell/material interactions and is one of the most important parameters which control the interactions.35 The initial cell adhesion on PLG scaffolds was evaluated after 12 h cultivation. As shown in Fig. 13, BMSCs adhered to all the surface of porous scaffolds. However, there were obvious differences in the cell attachment numbers and morphology on different samples. It was showed that small amount of cells adhered on the surface of PLA scaffold, which appeared small cell spreading area. What is more, the adhesion of BMSCs on the PLA scaffold treated with O.P showed no significant difference to the PLA samples. On the contrary, after 12 h adherent BMSCs in the composite scaffold showed increasing numbers and more prominent filopodia-like extensions. Furthermore, the number of BMSCs on the oxygen plasma-treated (O.P) PLG scaffolds was remarkably larger than the other samples and the cell spread better at this time point of culture. Previous researches have demonstrated that surface topography, wettability, porosity and pH value often play key roles in cell adhesion and cell spreading. Therefore, we hypothesized that the more hydrophilic PGS surface and desirable pH environment might contribute to the enhancement of BMSCs attachment on the PLG scaffolds. However, there were several green noises in the pictures of PLG samples. We believe that it may be attributed to the adsorption of green dye by PGS materials.
 | ||
| Fig. 13 Initial cell adhesion and spread arrangement of BMSCs after 12 h cultured onto the surface of PLA, PLA-O.P and PLG scaffolds. Representative fluorescent images with staining FITC-labeled phalloidin for cytoskeletal F-actin fibers (green) are shown (scale bar: 200 μm). | ||
The differentiation of BMSCs is very crucial to successful bone regeneration. We have determined the typical marker of osteoblast differentiation of ALP activity and the osteoblast markers of ALP, Runx2 and OCN. From Fig. 14(A), the ALP activities of the PLG samples greatly improved comparing with PLA scaffold and PLA-O.P scaffold after both 4 days and 7 days cell cultured. It was demonstrated that the existence of PGS might improve the expression of ALP activity and promote the differentiation of BMSCs. The gene expression of osteoblast markers were measured with real-time RT-PCR assay (Fig. 14(B)–(D)). The mRNA expression of ALP was increased significantly in BMSCs cultured with PLG groups. Especially, the PLG9 scaffold with oxygen plasma treatment showed the highest mRNA expression. Similar to the mRNA expression of ALP results, the gene expression of Runx2 (Fig. 14(C)) and OCN (Fig. 14(D)) also increased with the existence of PGS with PLA scaffold. Furthermore, the PLG9-O.P scaffold also displayed the highest Runx2 and OCN mRNA expression.
 | ||
| Fig. 14 Cell differentiation of BMSCs cultured with PLA, PLA-O.P and composite PLG scaffolds for 4 day and 7 days. (A) Alkaline phosphatase (ALP) activity of BMSCs cultured with different scaffolds. The expression of osteogenic marker gene (B) ALP, (C) Runx2 and (D) OCN determined by real-time RT-PCR analysis (n = 4, *p < 0.05). | ||
Collectively, PGS could effectively improve the mechanical strength, hydrophilicity and osteoconductivity and osteoinductivity of the conventional PLA scaffold for bone tissue regeneration, especially by the post-molding surface coating process with oxygen plasma pretreatment (O.P). Based on the previous investigations and the results presented here, an effective model was proposed to explore the reason involved in the various methods (Scheme 1).36 As discussed above, the mechanical strength of the final PGS/PLA scaffold largely depended on the combination of PLA and PGS. It is well accepted that the excellent elasticity of PGS results from the covalent crosslinking of the hydroxyl and carboxyl groups existing in pre-PGS through the esterification and hydrogen-bonding interactions between the hydroxyl groups attached to pre-PGS backbone during the curing process. For the pre-molding binary blend, PGS, as the minor phase, was distributed disorderly and discontinuously in the PLA. Therefore, it was hard for pre-PGS with a low molecular weight to form an effective and strong crosslinked network structure. While, surface coating of pre-PGS led to a homogeneous coating of PGS on the surface of porous PLA and thus resulted in a strong crosslinking reaction of PGS intermolecular reactions. Specifically, the oxygen-based plasma treatment could increase the oxygen-containing functional groups and the roughness of PLA surface, which are conducive to the spreading and conglutination of pre-PGS onto the surface of PLA and thus enhance the bonding strength of the pre-PGS or crosslinked PGS with the glossy original PLA surface. Therefore, the O.P-treated PLG/PLA scaffolds possessed the optimal interfacial and mechanical properties and thus exhibited enhanced osteoconductivity, osteoinductivity, biodegradability, and biomechanical properties, which can strongly widen the application and improve the properties of PLA in the field of bone tissue engineering scaffold.
 | ||
| Scheme 1 Schematic illustration of PLG scaffolds fabricated by B.B (i) and S.C (ii). Strengthening mechanism of oxygen-based plasma treatment modify the microtopography of PLA substrate and allow for a homogeneous coating of PGS tightly covered on the PLA surface (scale bar: 500 μm). | ||
Conclusions
PGS was used to modify PLA via pre-molding binary blend (B.B), and post-molding surface coating (S.C) without and with oxygen plasma pretreatment (D.C and O.P). The PLA and PLG scaffolds could be easily fabricated and presented well open-cell structure and connectivity. The results indicated that the addition of PGS to PLA improved the hydrophilicity, toughness and ductility and bioactivity of PLA scaffolds, especially for the oxygen plasma pretreatment group. Further studies demonstrated that effect of oxygen-base plasma treatment was due to the increased oxygen-containing functional groups and the roughened surface. PLG scaffolds prepared via O.P exhibited good biocompatibility and supported cell adhesion and proliferation. Furthermore, the PLG9-O.P scaffold displayed the highest osteogenic markers ALP, Runx2 and OCN mRNA expression in our research. Overall, this study indicated that the PGS could effectively modify the properties of PLA-based porous scaffolds and the fabricated PLA/PGS scaffolds could be promising clinical graft substitutes for bone and cartilage regeneration.Acknowledgements
The authors wish to express their gratitude for financial support from the National Basic Research Program of China (973 Program, No. 2012CB933600), National Natural Science Foundation of China (No. 31330028, No. 31470924, No. 31400817), the 111 Project (B14018), National Science and Technology Support Program (2014BAK05B02).Notes and references
- J. Zhang, H. Zhou, K. Yang, Y. Yuan and C. Liu, Biomaterials, 2013, 34, 9381–9392 CrossRef CAS PubMed.
- Y. Jung, S.-S. Kim, Y. H. Kim, S.-H. Kim, B.-S. Kim, S. Kim, C. Y. Choi and S. H. Kim, Biomaterials, 2005, 26, 6314–6322 CrossRef CAS PubMed.
- I. Ahmed, I. Jones, A. Parsons, J. Bernard, J. Farmer, C. Scotchford, G. Walker and C. Rudd, J. Mater. Sci.: Mater. Med., 2011, 22, 1825–1834 CrossRef CAS PubMed.
- R. A. Perez, K. D. Patel and H.-W. Kim, RSC Adv., 2015, 5, 13411–13419 RSC.
- A. Patel, A. K. Gaharwar, G. Iviglia, H. Zhang, S. Mukundan, S. M. Mihaila, D. Demarchi and A. Khademhosseini, Biomaterials, 2013, 34, 3970–3983 CrossRef CAS PubMed.
- W. Loued, J. Wéry, A. Dorlando and K. Alimi, J. Mol. Struct., 2015, 1081, 486–493 CrossRef CAS PubMed.
- A. Salerno, M. Fernández-Gutiérrez, J. S. R. del Barrio and C. Domingo, J. Supercrit. Fluids, 2015, 97, 238–246 CrossRef CAS PubMed.
- A. Salerno, M. Fernández-Gutiérrez, J. S. R. del Barrio and C. D. Pascual, RSC Adv., 2014, 4, 61491–61502 RSC.
- M. Frydrych and B. Chen, J. Mater. Chem. B, 2013, 1, 6650–6661 RSC.
- T. Saiga, S. Sato and K. Nagai, J. Appl. Polym. Sci., 2015, 132 DOI:10.1002/app.42200.
- C. E. LeBlon, R. Pai, C. R. Fodor, A. S. Golding, J. P. Coulter and S. S. Jedlicka, J. Appl. Polym. Sci., 2013, 128, 2701–2712 CrossRef CAS PubMed.
- S. Salehi, T. Bahners, J. Gutmann, S.-L. Gao, E. Mäder and T. Fuchsluger, RSC Adv., 2014, 4, 16951–16957 RSC.
- M. S. Mohammadi, I. Ahmed, N. Muja, C. D. Rudd, M. N. Bureau and S. N. Nazhat, J. Mater. Sci.: Mater. Med., 2011, 22, 2659–2672 CrossRef PubMed.
- Y. Wang, G. A. Ameer, B. J. Sheppard and R. Langer, Nat. Biotechnol., 2002, 20, 602–606 CrossRef CAS PubMed.
- Q. Z. Chen, J. M. Quinn, G. A. Thouas, X. Zhou and P. A. Komesaroff, Adv. Eng. Mater., 2010, 12, B642–B648 CrossRef PubMed.
- C. Zhu, S. R. Kustra and C. J. Bettinger, Acta Biomater., 2013, 9, 7362–7370 CrossRef CAS PubMed.
- Z.-J. Sun, L. Wu, X.-L. Lu, Z.-X. Meng, Y.-F. Zheng and D.-L. Dong, Appl. Surf. Sci., 2008, 255, 350–352 CrossRef CAS PubMed.
- S.-L. Liang, W. D. Cook, G. A. Thouas and Q.-Z. Chen, Biomaterials, 2010, 31, 8516–8529 CrossRef CAS PubMed.
- B. Xu, B. Rollo, L. A. Stamp, D. Zhang, X. Fang, D. F. Newgreen and Q. Chen, Biomaterials, 2013, 34, 6306–6317 CrossRef CAS PubMed.
- Q. Chen, S. Liang and G. A. Thouas, Soft Matter, 2011, 7, 6484–6492 RSC.
- S.-L. Liang, X.-Y. Yang, X.-Y. Fang, W. D. Cook, G. A. Thouas and Q.-Z. Chen, Biomaterials, 2011, 32, 8486–8496 CrossRef CAS PubMed.
- I. H. Jaafar, C. E. LeBlon, M.-T. Wei, D. Ou-Yang, J. P. Coulter and S. S. Jedlicka, Acta Biomater., 2011, 7, 1588–1598 CrossRef CAS PubMed.
- C. L. Nijst, J. P. Bruggeman, J. M. Karp, L. Ferreira, A. Zumbuehl, C. J. Bettinger and R. Langer, Biomacromolecules, 2007, 8, 3067–3073 CrossRef CAS PubMed.
- V. T. Phuong, M.-B. Coltelli, P. Cinelli, M. Cifelli, S. Verstichel and A. Lazzeri, Polymer, 2014, 55, 4498–4513 CrossRef CAS PubMed.
- H. Yu, C. Yan and J. Yao, RSC Adv., 2014, 4, 59792–59802 RSC.
- Y. B. Kim and G. Kim, J. Mater. Chem., 2012, 22, 16880–16889 RSC.
- L. Zhou, H. He, C. Jiang and S. He, J. Appl. Polym. Sci., 2015, 132 DOI:10.1002/app.42196.
- N. De Geyter, R. Morent, T. Desmet, M. Trentesaux, L. Gengembre, P. Dubruel, C. Leys and E. Payen, Surf. Coat. Technol., 2010, 204, 3272–3279 CrossRef CAS PubMed.
- H. Yu, Z. Z. Chong, S. B. Tor, E. Liu and N. H. Loh, RSC Adv., 2015, 5, 8377–8388 RSC.
- P. Dixit and J. Miao, J. Electrochem. Soc., 2006, 153, G771–G777 CrossRef CAS PubMed.
- C. Alves, Y. Yang, D. Marton, D. Carnes, J. Ong, V. Sylvia, D. Dean, R. Reis and C. Agrawal, J. Biomed. Mater. Res., Part B, 2008, 87, 59–66 CrossRef CAS PubMed.
- E. Tejeda-Montes, A. Klymov, M. R. Nejadnik, M. Alonso, J. C. Rodriguez-Cabello, X. F. Walboomers and A. Mata, Biomaterials, 2014, 35, 8339–8347 CrossRef CAS PubMed.
- D. Ju, L. Han, J. Bian, Z. Guo, F. Li, S. Chen and L. Dong, RSC Adv., 2015, 5, 5474–5483 RSC.
- T. H. Qazi, R. Rai, D. Dippold, J. E. Roether, D. W. Schubert, E. Rosellini, N. Barbani and A. R. Boccaccini, Acta Biomater., 2014, 10, 2434–2445 CrossRef CAS PubMed.
- Q. Gan, X. Lu, Y. Yuan, J. Qian, H. Zhou, X. Lu, J. Shi and C. Liu, Biomaterials, 2011, 32, 1932–1942 CrossRef CAS PubMed.
- A. Boularesres, Y. Yuan, J. Qian, H. Zhou, X. Lu, J. Shi and R. Buchmeiser, J. Appl. Polym. Sci., 2011, 121, 2543–2550 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |