
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA concise route to functionalized benzofurans directly from gem-dibromoalkenes and phenols†
Maddali L. N.
Rao
* and
Priyabrata
Dasgupta
Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur, India. E-mail: maddali@iitk.ac.in; Fax: +91-512-2597532
First published on 31st July 2015
Abstract
A tandem strategy for the construction of benzofuran motifs has been developed directly from gem-dibromoalkenes and phenols under palladium-catalyzed conditions. This flexible and novel methodology provides direct access to 2-aryl and 2-styryl benzofurans in good to high yields. This strategy is also valuable in the synthesis of benzodifurans.
Introduction
Methods involving gem-dibromoalkenes (1,1-dibromoalkenes) as synthetic surrogates are of immense interest with ready availability as molecular synthons in organic synthesis.1 Reactions involving gem-dibromoalkenes and o-hydroxy-gem-dibromoarylalkenes in combination with triarylbismuth reagents have been reported from this laboratory for the preparation of internal alkynes2a and 2-arylbenzofurans2b respectively under palladium-catalyzed conditions. There are other applications of gem-dibromoalkenes3 in organic synthesis as reviewed by Chellucci et al. recently.1 To note, benzofurans are medicinal scaffolds with wide pharmacological appeal and presence in natural products.4 Synthesis of these skeletons through sustainable approach and in one-pot operation allows minimization of waste, time and effort together with an easy execution of the target reaction protocol.5Hence, we envisioned the pot-economic strategy as ideal choice for the synthesis of 2-arylbenzofuran scaffold through direct involvement of 1,1-dibromoalkene and phenol as given in Scheme 1. This approach (i) would alleviate the use of organometallic reagent as coupling partner,3a,b (ii) would be practicable as the proposed 1,1-dibromoalkene could be derived from aldehydes,6 (iii) is simpler in terms of overall substrate combination and (iv) provides a wide synthetic and functional scope with 1,1-dibromoalkene as a reactant. Further, the starting materials phenols and aldehydes are routinely available. Additionally, this approach utilizes in situ preparation and functionalization of more reactive and not so stable 1-bromoalkyne without any isolation.
 | ||
| Scheme 1 One-pot strategy. | ||
That way, the present approach is more advantageous from the fact that 1-bromoalkynes are photolabile and cannot be used/accessed as bench top chemicals.7 The most common method of preparation of 1-bromoalkynes relies on the bromination of terminal alkynes with either CBr4/PPh3 (ref. 8) or Ag(I)/NBS9 system. The proposed overall strategy is thus expected to provide an easy access to variously functionalized benzofurans with multiple practical and synthetic benefits.
Procedurally, the generation of benzofuran would thus involves (i) initial elimination,2a (ii) addition reaction sequence for the generation of 2-bromovinyl aryl ether and (iii) its oxidative cyclization to generate benzofuran in a one-pot operation (vide infra).10 However, the overall outcome practically depends on how effectively these three steps could be converged under a viable protocol with high synthetic utility for the preparation of benzofurans.
Results and discussion
We studied our strategy with direct use of 1,1-dibromoalkene (1a) and phenol (1b) as model substrates for Step 1 to generate 2-bromovinyl phenyl ether (1c). This was explored using different combinations of base, solvent, temperature and reaction time conditions (Table 1).|
|
|||||
|---|---|---|---|---|---|
| Entry | Base (equiv.) | Solvent | Temp (°C) | Time (h) | 1c (%) |
| a Conditions: 1-(2,2-dibromovinyl)-4-methylbenzene, 1a (0.375 mmol, 1 equiv.), 4-nitrophenol, 1b (0.375 mmol, 1 equiv.), base, solvent (3 mL), temp, time. b Isolated yields. | |||||
| 1 | K3PO4 (3) | DMF | 90 | 4 | 16 |
| 2 | K3PO4 (3) | DMF | 110 | 8 | 70 |
| 3 | K3PO4 (3) | NMP | 110 | 8 | 75 |
| 4 | KOAc (3) | NMP | 110 | 8 | 32 |
| 5 | K2CO3 (3) | NMP | 110 | 8 | 57 |
| 6 | Cs2CO3 (3) | NMP | 110 | 8 | 78 |
| 7 | Cs2CO3 (5) | NMP | 110 | 8 | 87 |
| 8 | Cs2CO3 (5) | DMA | 110 | 8 | 73 |
| 9 | Cs2CO3 (5) | DMF | 110 | 8 | 67 |
| 10 | Cs2CO3 (5) | NMP | 110 | 6 | 79 |

The initial examination of this reaction with K3PO4 base at 90 °C in DMF for 4 h afforded 2-bromovinyl phenyl ether (1c) in 16% yield (entry 1, Table 1). However, notable improvement was achieved with this base at 110 °C both in DMF and NMP solvents giving 70% and 75% yields respectively (entries 2 and 3, Table 1). Replacing K3PO4 either with KOAc or K2CO3 proved to be ineffective with lowered yields (entries 4 and 5, Table 1). Encouragingly, this reaction in NMP with Cs2CO3 (3 or 5 equiv.) demonstrated with high performance giving 78% and 87% yields (entries 6 and 7, Table 1). Further, change in solvent to either DMA or DMF resulted in lowered yields (entries 8 and 9, Table 1). Further screening with 6 h time duration furnished 79% yield (entry 10, Table 1). This investigation using different combinations of base, solvent, temperature and time conditions indicated that the formation of 2-bromovinyl phenyl ether (1c) is efficient with Cs2CO3 (5 equiv.) in NMP to give high yield (entry 7, Table 1). Considering this as optimized condition for Step 1, further attempt was made to converge the protocol for Step 1 with the proposed cyclization in Step 2 under palladium catalysis towards benzofuran formation. The underlying challenge here is in finding the compatible palladium conditions in convergence with the conditions of Step 1. Thus, the oxidative cyclization of 2-bromovinyl phenyl ether (1c) should be affected under palladium catalysis within the protocol limitation of Step 1.
With the above prerequisite, the screening for Step 2 was performed using different palladium catalysts under the protocol conditions of Step 1 (Table 2). This investigation initially carried out with PdCl2 or PdCl2(PPh3)2 at 130 °C furnished the desired benzofuran product 2.1 in moderate yields (entries 1 and 2, Table 2). To our satisfaction, the desired cyclization improved up to 78% using with Pd(OAc)2 (entry 3, Table 2). Further check at 110 °C or with lowered amount of catalyst gave 70% (entry 4, Table 2) and 61% (entry 5, Table 2) yields respectively. It was realized that the cyclization of 2-bromovinyl phenyl ether (1c) could be achieved with Pd(OAc)2 under the conditions of Step 1 in a two-step one-pot operation with high yield (entry 3, Table 2). It is to be highlighted that the formation of 2-bromovinyl phenyl ether (1c) from 1,1-dibromide (1a) and phenol (1b) was achieved efficiently with Cs2CO3 in NMP at 110 °C in Step 1 followed by heating 130 °C in the presence of Pd(OAc)2 to afford 2-arylbenzofuran 2.1 directly in high yield through a one-pot operation (entry 3, Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (equiv.) | Temp (°C) | Time (h) | 2.1 (%) |
| a Conditions: Step 1: 1a (0.375 mmol, 1 equiv.), 1b (0.375 mmol, 1 equiv.), Cs2CO3 (1.875 mmol, 5 equiv.), NMP (3 mL), 110 °C, 8 h; Step 2: [Pd] catalyst, temp, time. b Isolated yields refer to overall yields after two steps. | ||||
| 1 | PdCl2 (0.05) | 130 | 6 | 54 |
| 2 | PdCl2(PPh3)2 (0.05) | 130 | 6 | 45 |
| 3 | Pd(OAc)2 (0.05) | 130 | 6 | 78 |
| 4 | Pd(OAc)2 (0.05) | 110 | 6 | 70 |
| 5 | Pd(OAc)2 (0.03) | 130 | 6 | 61 |

This one-pot direct strategy using 1,1-dibromoalkene and phenol is synthetically more viable for the preparation of a variety of 2-arylbenzofurans. Thus, further investigation was carried out to expand the scope using different functionalized 1,1-dibromides and phenols as summarized in Table 3. The reactivity of 1-(2,2-dibromovinyl)-4-methylbenzene was tested initially with various electronically different phenols. The study with 4-nitrophenol during screening afforded 5-nitro-2-(p-tolyl)benzofuran, 2.1 in 78% yield. This reactivity with different phenols functionalized with 4-cyano-, 4-fluoro-, 4-chloro-, 2,4-dichloro groups afforded the corresponding benzofurans (2.2–2.5) in 55–67% yields. Similarly, 1-naphthol efficiently furnished benzofuran 2.6 in 66% yield. Further, electronically rich phenols substituted with 4-methyl, 2,3-dimethyl, 3,5-dimethyl groups provided benzofurans (2.7–2.9) in 51–57% yields. Notably, simple phenol gave benzofuran 2.10 in 51% yield. Comparable reactivity was witnessed with unsubstituted (2,2-dibromovinyl)benzene in combination with different phenols to deliver the corresponding benzofurans (2.11–2.14) in 56–68% yields. The study of 4-chloro and 4-methoxy substituted (2,2-dibromovinyl)benzenes demonstrated persistent reactivity and gave benzofurans (2.15–2.22) in 47–78% yields. Reaction with 2-chlorophenol afforded benzofuran 2.23 in 55% yield. Reactivity with 3-chloro and 3-nitro substituted phenols provided benzofurans 2.24 and 2.25 in 53% and 63% yields respectively. Further ester functionalized phenolic substrate such as methyl 4-hydroxybenzoate delivered benzofuran 2.26 in 77% high yield. The same with 3-chloro and 4-fluoro substituted (2,2-dibromovinyl)benzenes provided benzofurans 2.27 and 2.28 in moderate yields. Thus, moderate yields were obtained in reactions with electron rich phenols. In some of these cases, we noticed the formation of bis-phenoxy intermediates through mass spectral analysis in minor amounts.
|
|
|---|
| a Conditions: Step 1: 1,1-dibromide (0.375 mmol, 1 equiv.), phenol (0.375 mmol, 1 equiv.), Cs2CO3 (1.875 mmol, 5 equiv.), NMP, 110 °C, 8 h; Step 2: Pd(OAc)2 (0.0187 mmol, 0.05 equiv.), 130 °C, 6 h. b Isolated yields refer to overall yields after two steps in one-pot operation and corresponds to 0.375 mmol of product as 100% yield. |

|

This exploration with different 1,1-dibromides and phenols demonstrated the overall synthetic potential of the developed direct strategy and was driven by electronics of both dibromides and phenols. In fact, considerable electronic influence was observed both in the Step 1 and Step 2 which led to overall moderate to good yields in one-pot operation.
Notably, some of the 2-arylbenzofuran skeletons obtained are useful intermediates with structural modifications in the preparation of natural products.4 Additionally, benzofuran 2.21 could be modified for the synthesis of a β-amyloid aggregation inhibitor.4f
At this stage, it was further decided to exploit the advantage and flexibility available with our system to achieve other functionalizations at C-2 position of benzofuran employing differently functionalized 1,1-dibromides. We envisaged one such possibility with the use of 1,3-dienyldibromide to synthesize 2-styryl substituted benzofuran products (Scheme 1). In fact, synthesis of these skeletons through earlier procedures (Scheme 1) require the use of styryl based organometallic reagents which is a cumbersome in terms of preparation and other related issues.11 Whereas, 1,3-dienyldibromides could be readily obtained from cinnamaldehydes and their direct use in comparison is expected to provide immense synthetic utility (Scheme 1). With this advantage in hand, we briefly reviewed this possibility using 1,3-dienyldibromide in the preparation of 2-styrylbenzofuran (Table 4).
|
|
|---|
| a Conditions: Step 1: 1,3-dienyldibromide (0.375 mmol, 1 equiv.), phenol (0.375 mmol, 1 equiv.), Cs2CO3 (1.875 mmol, 5 equiv.), NMP, 110 °C, 8 h; Step 2: Pd(OAc)2 (0.0187 mmol, 0.05 equiv.), 130 °C, 6 h. b Isolated yields refer to overall yields after two steps in one-pot corresponds to 0.375 mmol of product as 100% yield. |

|
Amazingly, our attempt afforded 2-styrylbenzofurans in a facile manner using above established conditions. This effort also ascertained the synthetic advantage as differently functionalized 2-styrylbenzofurans (3.1–3.4) could be obtained directly in 48–60% yields. To note, 2-styrylbenzofurans are valuable skeletons for biological and other applications.4g,h Given the lack of readily usable methods for the preparation of these products directly,12 the present method is significant employing 1,3-dienyldibromides.
Further, to investigate and converge the reaction course in line with the screening carried out in Table 1 involving the isolation of the intermediate, an additional experimentation was desired with 1,3-dienyldibromide. Thus was performed directly under the conditions of Step 1 and the corresponding elimination–addition product 3c was isolated in 73% yield (Scheme 2).
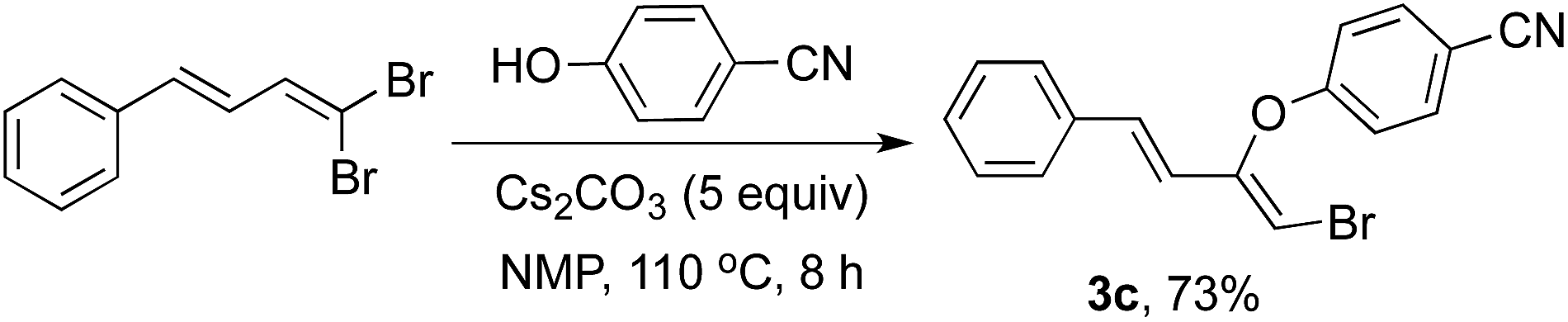 | ||
| Scheme 2 Isolation of the reaction intermediate 3c. | ||
Synthesis of important skeletons like benzodifurans has always been challenging pursuit.4i,j Hence, it was attempted here with dibromides and resorcinols in a stepwise manner with the isolation of the intermediate structures. In this attempt the reaction of 1a with simple resorcinol gave intermediate 4c in 50% yield as isomeric mixture. When 4c further subjected for cyclization under Pd(OAc)2/Cs2CO3 conditions, it delivered benzodifuran 4.1 in 48% yield (Scheme 3). Similar attempt with 4-methoxy substituted (2,2-dibromovinyl)benzene and methyl 3,5-dihydroxybenzoate gave benzodifuran 5.1 in 52% yield involving 5c (Scheme 3). This study in one-pot operation although failed to deliver benzodifuran, stepwise approach successfully delivered benzodifurans in moderate yields involving tandem process.
 | ||
| Scheme 3 Stepwise synthesis of benzodifurans 4.1 and 5.1. | ||
The single crystal X-ray analysis of 4.1 (Fig. 1) showed the double cyclizations C-3 and C-9 leading to benzodifuran cyclized product with bowl shaped fused tricyclic core.
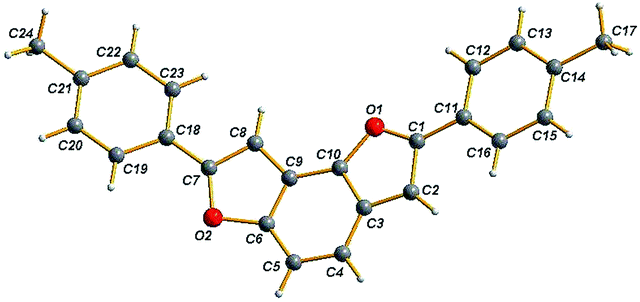 | ||
| Fig. 1 X-ray structure of 4.1. | ||
Additional efforts to apply our protocol to gram scale reaction of dibromide 1a in combination with methyl 4-hydroxybenzoate in a two-step one-pot operation delivered 2-arylbenzofuran 2.26 in 42% yield (Scheme 4).
 | ||
| Scheme 4 Gram scale reaction. | ||
The mechanistic pathway for the formation of benzofuran from 1,1-dibromide and phenol is given Scheme 5.
 | ||
| Scheme 5 Mechanistic proposal. | ||
During Step 1, the initial dehydrobromination of 1,1-dibromide in the presence of base would form 1-bromoalkyne (3p). This in turn undergoes addition reaction with phenol to afford 2-bromovinyl phenyl ether (3q).10,12d,13 In Step 2, 2-bromovinyl phenyl ether (3q) would undergo a sequence of steps involving oxidative addition (3r), cyclization (3s) followed by reductive elimination to generate 2-substituted benzofuran in one-pot operation. The formation of 1-bromoalkyne in the presence of base was realized on our earlier studies both in the case of 1,1-dibromoalkene2a and 1,3-dienyldibromide.14 Further it was evidenced by the isolation of the addition adducts 1c (Table 1) and 3c (Scheme 2). The second step of oxidative cyclization under palladium-catalyzed conditions is well represented in the literature towards benzofuran formation.10,12d,13
It is to be emphasized that with readily available starting materials, the present approach offers an easy access to the 2-aryl and 2-styryl substituted benzofurans without using organometallic coupling reagents such as aryl and alkenylboronic acids. This is notable as alkenylboronic acids reagents suffer from unavoidable practical difficulties.11,15 and our method is a devoid of these short comings in the preparation of benzofurans.
Conclusion
In conclusion, a direct tandem strategy for the construction of 2-aryl and 2-styrylbenzofuran motifs has been developed from 1,1-dibromides and phenols under palladium-catalyzed conditions. This flexible and novel methodology provides an easy access to functionalized benzofurans in a pot-economic practical way. Our study is also worthwhile to access benzodifurans in a facile manner.Experimental
General
All reactions were conducted in Schlenk tubes under nitrogen atmosphere with dry solvents. Chromatographic purification of the products were performed using 100–200 and 230–400 mesh silica-gel with ethyl acetate/hexane as eluent. NMR spectra have been recorded using JEOL ECS 400 (400 MHz) and JEOL ECX 500 (500 MHz). HRMS measurements have been done using Electron Ionization (EI) and Electrospray Ionization (ESI) techniques with Waters CAB155 GCT Premier analyzer and Waters HAB 213 Q-TOF Premier analyzer. IR spectra were recorded with PerkinElmer FT-IR Spectrum Two and Bruker Tensor 27 spectrometer. X-ray data was recorded on Bruker SMART APEX-II CCD diffractometer. Different gem-dibromoalkenes were prepared according to reported procedures.2,14 Melting points reported are uncorrected.Representative procedure for intermediate 1c and 3c
This reaction was performed using an oven-dried Schlenk tube charged with 1-(2,2-dibromovinyl)-4-methylbenzene 1a (0.104 g, 0.375 mmol, p-nitrophenol 1b (0.52 g, 0.375 mmol, 1 equiv.), 1 equiv.), Cs2CO3 (0.611 g, 1.875 mmol, 5 equiv.) and NMP (3 mL) respectively under N2 atmosphere. Reaction mixture was stirred at 110 °C in an oil bath for 8 h. It was cooled to rt and subjected to workup using ethyl acetate (50 mL), washing with water (10 mL), brine (10 mL). The organic portion was dried over anhydrous MgSO4 and concentrated. The crude was purified by silica-gel column chromatography using 2% EtOAc in hexane as eluent to obtain the product 1c as yellow viscous liquid (0.109 g, 87%). This procedure was also followed for the preparation of intermediate 3c with (E)-(4,4-dibromobuta-1,3-dienyl)benzene and p-cyanophenol.Representative procedure for 2-arylbenzofurans (Table 3)
This reaction was performed in an oven-dried Schlenk tube with dibromide 1a (0.104 g, 0.375 mmol, 1 equiv.), 1b (0.52 g, 0.375 mmol, 1 equiv.), Cs2CO3 (0.611 g, 1.875 mmol, 5 equiv.) and NMP (3 mL) under N2 atmospheric conditions. Reaction mixture was stirred at 110 °C in an oil bath for 8 h. It was cooled to rt for the addition of Pd(OAc)2 (0.0042 g, 0.0187 mmol, 0.05 equiv.) and reaction was further heated at 130 °C for 6 h under N2 atmosphere conditions. It was cooled to rt and was extracted with ethyl acetate (50 mL), washed with water (10 mL), brine (10 mL), dried over anhydrous MgSO4 and concentrated. The crude was subjected to silica gel column chromatography using 2% EtOAc in hexane as eluent to obtain the product 5-nitro-2-(p-tolyl)benzofuran (2.1) as brown solid (0.074 g, 78%).Representative procedure for 2-styrylbenzofurans (Table 4)
The procedure followed above in the preparation of benzofuran 2.1 was adopted with 1,3-dienyldibromides and phenols for the synthesis of 2-styrylbenzofurans 3.1–3.4.Representative procedure for benzodifurans (4.1 and 5.1)
It was performed in stepwise manner in an oven-dried Schlenk tube with dibromide 1a (0.31 g, 1.125 mmol, 3 equiv.), resorcinol (0.041 g, 0.375 mmol, 1 equiv.), Cs2CO3 (1.22 g, 3.75 mmol, 10 equiv.) and NMP (3 mL) under N2 atmospheric conditions. The reaction mixture was stirred at 110 °C in an oil bath for 8 h. After usual workup and purification using silica-gel column chromatography with 5% EtOAc in hexane 4c was obtained as viscous liquid (0.095 g, 50%). The compound 4c (0.095 g, 0.19 mmol, 1 equiv.) was subjected for cyclization using conditions Cs2CO3 (0.25 g, 0.76 mmol, 4 equiv.), Pd(OAc)2 (0.0042 g, 0.019 mmol, 0.10 equiv.) and NMP (2 mL) under N2 and heated at 130 °C for 6 h in the second step. After work up and purification as above, benzodifuran 4.1 was obtained as colourless solid (0.031 g, 48%). Similarly, benzodifuran 5.1 was obtained in 52% yield from 0.142 mmol of intermediate 5c. For analytical data of intermediates 4c and 5c, see ESI.†The analytical data for intermediates 1c, 3c, benzofuran products 2.1–2.28, 3.1–3.4, 4.1 and 5.1 are given below.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95). 1H NMR (400 MHz, CDCl3): δ 8.17 (d, 2H, J = 9.2 Hz, Ar–H), 7.34 (d, 2H, J = 8.24 Hz, Ar–H), 7.14 (d, 2H, J = 8 Hz, Ar–H), 7.07–7.03 (m, 2H, Ar–H), 6.58 (s, 1H, C
:95). 1H NMR (400 MHz, CDCl3): δ 8.17 (d, 2H, J = 9.2 Hz, Ar–H), 7.34 (d, 2H, J = 8.24 Hz, Ar–H), 7.14 (d, 2H, J = 8 Hz, Ar–H), 7.07–7.03 (m, 2H, Ar–H), 6.58 (s, 1H, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) olefin), 2.32 (s, 3H, –C3). 13C (125 MHz, CDCl3): δ 160.8, 152.5, 142.7, 140.1, 129.8, 129.6, 126.0, 125.4, 116.0, 95.4, 21.3. IR (film): 3093, 2923, 1610, 1590, 1517, 1343, 1238, 1111 cm−1. HRMS (EI) calcd for C15H12BrNO3 [M+] 333.0001; found 333.0009.
:95). 1H NMR (400 MHz, CDCl3): δ 7.61 (d, 2H, J = 9.16 Hz, Ar–H), 7.36–7.25 (m, 5H, Ar–H), 7.08 (d, 2H, J = 9.16 Hz, Ar–H), 6.73–6.64 (m, 2H), 6.34 (s, 1H, Colefin). 13C (100 MHz, CDCl3): δ 159.3, 151.9, 135.3, 134.2, 131.6, 128.8, 126.9, 119.7, 118.8, 116.0, 105.8, 99.9. IR (film): 3081, 2226, 1684, 1608, 1502, 1236, 1168 cm−1. HRMS (EI) calcd for C17H12BrNO [M+] 325.0102; found 325.0105.
:98). 1H NMR (400 MHz, CDCl3): δ 8.48 (d, 1H, J = 2.28 Hz, Ar–H), 8.20 (dd, 1H, J = 8.9 Hz, J = 2.52 Hz, Ar–H), 7.77 (d, 2H, J = 8.24 Hz, Ar–H), 7.57 (d, 1H, J = 9.16 Hz, Ar–H), 7.29 (d, 2H, J = 8.24 Hz, Ar–H), 7.06 (s, 1H, Ar–H), 2.42 (s, 3H, –C3). 13C (100 MHz, CDCl3): δ 159.5, 157.5, 144.2, 139.9, 129.7, 126.4, 125.2, 119.8, 117.0, 111.3, 100.8, 21.4. IR (film, cm−1): 2920, 1589, 1519, 1504, 1342, 886, 792, 749. HRMS (ESI) calcd for C15H12NO3 [MH+] 254.0817; found 254.0815.
:95). 1H NMR (400 MHz, CDCl3): δ 7.88 (s, 1H, Ar–H), 7.74 (d, 2H, J = 8.24 Hz, Ar–H), 7.57–7.51 (m, 2H, Ar–H), 7.28–7.24 (m, 2H, Ar–H), 6.98 (s, 1H, Ar–H), 2.40 (s, 3H, –C3). 13C (100 MHz, CDCl3): δ 158.6, 156.3, 139.8, 130.0, 129.6, 127.6, 126.5, 125.5, 125.2, 119.5, 112.1, 106.7, 99.9, 21.4. IR (film): 2923, 2225, 1589, 1463, 1032, 821, 798 cm−1. HRMS (EI) calcd for C16H11NO [M+] 233.0841; found 233.0843.
3). 13C (125 MHz, CDCl3): δ 159.3 (d, J = 236.37 Hz), 157.9, 150.9, 139.0, 130.1 (d, J = 10.8 Hz), 129.5, 127.3, 124.9, 111.6, 111.5, 111.4, 106.1 (d, J = 25.2 Hz), 100.6 (J = 3.6 Hz), 21.4. IR (film): 2961, 1724, 1590, 1461, 1272, 1112, 864, 822, 796 cm−1. HRMS (EI) calcd for C15H11FO [M+] 226.0794; found 226.0799.
3). 13C (125 MHz, CDCl3): δ 157.6, 153.1, 139.1, 130.7, 129.5, 128.3, 127.2, 124.9, 124.0, 120.2, 111.9, 100.0, 21.4. IR (film): 2961, 1725, 1446, 1273, 1035, 824, 808, 796 cm−1. HRMS (EI) calcd for C15H11ClO [M+] 242.0498; found 242.0492.
3). 13C (125 MHz, CDCl3): δ 158.4, 149.0, 139.6, 131.6, 129.5, 128.5, 126.5, 125.1, 123.9, 118.8, 116.9, 100.4, 21.4. IR (film): 2960, 1724, 1575, 1505, 1443, 1277, 1169, 835, 817, 793 cm−1. HRMS (EI) calcd for C15H10Cl2O [M+] 276.0109; found 276.0104.
3). 13C (100 MHz, CDCl3): δ 155.6, 150.1, 138.2, 131.3, 129.5, 128.4, 128.0, 126.3, 124.9, 124.6, 123.5, 121.3, 120.0, 119.5, 101.7, 21.4. IR (film): 3052, 1498, 915, 817, 743 cm−1. HRMS (EI) calcd for C19H14O [M]+ 258.1045; found 258.1046.
3), 2.39 (s, 3H, –C3). 13C (125 MHz, CDCl3): δ 156.2, 153.2, 138.4, 132.2, 129.4, 127.9, 125.2, 124.8, 120.5, 110.5, 100.3, 21.36, 21.33. IR (film): 2962, 1726, 1465, 1289, 823, 800 cm−1. HRMS (EI) calcd for C16H14O [M+] 222.1045; found 222.1043.
3), 2.39 (s, 6H, –C3). 13C (125 MHz, CDCl3): δ 155.3, 154.2, 138.1, 132.4, 129.4, 128.1, 126.6, 124.9, 124.6, 119.6, 117.2, 100.8, 21.3, 19.2, 11.7. IR (film): 3029, 2920, 1726, 1505, 1288, 814 cm−1. HRMS (EI) calcd for C17H16O [M+] 236.1201; found 236.1209.
3), 2.44 (s, 3H, –C3), 2.39 (s, 3H, –C3). 13C (125 MHz, CDCl3): δ 155.0, 154.9, 138.1, 134.2, 130.1, 129.4, 128.0, 126.6, 124.6, 124.5, 108.7, 99.1, 21.7, 21.3, 18.5. IR (film): 3024, 2919, 1727, 1619, 1504, 1294, 1036, 836, 820, 794 cm−1. HRMS (EI) calcd for C17H16O [M+] 236.1201; found 236.1209.
3). 13C (125 MHz, CDCl3): δ 156.1, 154.7, 138.6, 129.4, 129.3, 127.7, 124.8, 123.9, 122.8, 120.7, 111.0, 100.5, 21.4. IR (film): 2932, 1725, 1452, 1270, 801, 749, 737 cm−1. HRMS (EI) calcd for C15H12O [M+] 208.0888; found 208.0880.
:95). 1H NMR (500 MHz, CDCl3): δ 7.91 (s, 1H, Ar–H), 7.87 (d, 2H, J = 7.3 Hz, Ar–H), 7.60–7.54 (m, 2H, Ar–H), 7.48 (t, 2H, J = 7.5 Hz, Ar–H), 7.43–7.40 (m, 1H, Ar–H), 7.05 (s, 1H, Ar–H). 13C (125 MHz, CDCl3): δ 158.3, 156.4, 129.8, 129.5, 129.2, 128.9, 127.8, 125.7, 125.2, 119.4, 112.2, 106.8, 100.7. IR (film): 3093, 2960, 2226, 1723, 1465, 1288, 896, 825, 747, 766 cm−1. HRMS (ESI) calcd for C15H10NO [MH+] 220.0762; found 220.0769.
:95). 1H NMR (500 MHz, CDCl3): δ 7.90 (s, 1H, Ar–H), 7.78 (d, 2H, J = 8.6 Hz, Ar–H), 7.58–7.54 (m, 2H, Ar–H), 7.44 (d, 2H, J = 8.05 Hz, Ar–H), 7.02 (s, 1H, Ar–H). 13C (125 MHz, CDCl3): δ 157.1, 156.4, 135.4, 129.7, 129.2, 128.1, 127.7, 126.4, 125.8, 119.3, 112.3, 107.0, 101.1. IR (film): 2960, 2225, 1723, 1486, 1461, 1269, 1090, 810, 793 cm−1. HRMS (EI) calcd for C15H8ClNO [M+] 253.0294; found 253.0290.
:98). 1H NMR (500 MHz, CDCl3): δ 8.51 (s, 1H, Ar–H), 8.23 (d, 1H, J = 9.2 Hz, Ar–H), 7.81 (d, 2H, J = 8 Hz, Ar–H), 7.59 (d, 1H, J = 9.2 Hz, Ar–H), 7.47 (d, 2H, J = 8 Hz, Ar–H), 7.12 (s, 1H, Ar–H). 13C (125 MHz, CDCl3): δ 158.1, 157.6, 144.4, 135.6, 129.5, 129.3, 127.7, 126.5, 120.3, 117.4, 111.5, 102.0. IR (film): 3099, 2961, 1722, 1588, 1520, 1486, 1348, 1268, 1090, 793 cm−1. HRMS (EI) calcd for C14H8ClNO3 [M+] 273.0193; found 273.0197.
3), 2.40 (s, 3H, –C3). 13C (125 MHz, CDCl3): δ 154.4, 153.9, 133.8, 133.0, 129.3, 128.9, 126.3, 125.9, 125.1, 119.8, 117.5, 102.0, 19.3, 11.7. IR (film): 3037, 2921, 1725, 1481, 1090, 814, 750 cm−1. HRMS (EI) calcd for C16H13ClO [M+] 256.0655; found 256.0650.
3). 13C (100 MHz, CDCl3): δ 159.7, 155.4, 149.9, 131.2, 128.4, 126.2, 126.1, 125.0, 124.7, 123.6, 123.4, 121.3, 119.9, 119.4, 114.3, 100.8, 55.3. IR (film): 2926, 1500, 1252, 1032, 814, 748 cm−1. HRMS (EI) calcd for C19H14O2 [M]+ 274.0994; found 274.0985.
:98). 1H NMR (400 MHz, CDCl3): δ 7.77 (d, 2H, J = 9.16 Hz, Ar–H), 7.50 (d, 1H, J = 1.84 Hz, Ar–H), 7.40 (d, 1H, J = 8.68 Hz, Ar–H), 7.19 (dd, 1H, J = 8.68 Hz, J = 1.84 Hz, Ar–H), 6.98 (d, 2H, J = 8.68 Hz, Ar–H), 6.81 (s, 1H, Ar–H), 3.86 (s, 3H, –OC3). 13C (100 MHz, CDCl3): δ 160.3, 157.5, 153.0, 130.8, 128.3, 126.5, 123.8, 122.7, 120.0, 114.3, 111.9, 99.1, 55.3. IR (film): 2964, 1724, 1505, 1271, 1040, 832, 796 cm−1. HRMS (EI) calcd for C15H11ClO2 [M+] 258.0448; found 258.0445.
:98). 1H NMR (500 MHz, CDCl3): δ 7.79 (d, 2H, J = 6.85 Hz, Ar–H), 7.39 (s, 1H, Ar–H), 7.25–7.23 (m, 1H, Ar–H), 6.97 (d, 2H, J = 7.45 Hz, Ar–H), 6.81 (s, 1H, Ar–H), 3.86 (s, 3H, –OC3). 13C (125 MHz, CDCl3): δ 160.6, 158.3, 149.0, 131.8, 128.5, 126.8, 123.7, 122.1, 118.7, 116.9, 114.3, 99.5, 55.4. IR (film): 2967, 1720, 1610, 1505, 1445, 1255, 824, 792 cm−1. HRMS (EI) calcd for C15H10Cl2O2 [M+] 292.0058; found 292.0050.
3). 13C (100 MHz, CDCl3): δ 157.0, 150.5, 139.1, 130.9, 129.5, 127.1, 125.1, 124.1, 123.7, 119.2, 116.5, 100.9, 21.4. IR (film): 3033, 1589, 1471, 1193, 1034, 905, 805, 734 cm−1. HRMS (EI) calcd for C15H11ClO [M+] 242.0498; found 242.0497.
3). 13C (100 MHz, CDCl3): δ 157.0, 154.8, 138.9, 129.5, 128.0, 127.3, 124.9, 123.6, 121.1, 111.6, 100.2, 21.4. IR (film): 3092, 1582, 1503, 1423, 1030, 925, 816 cm−1. HRMS (EI) calcd for C15H11ClO [M+] 242.0498; found 242.0495.
:9).1H NMR (400 MHz, CDCl3): δ 8.40 (s, 1H, Ar–H), 8.17 (dd, 1H, J = 8.72 Hz, J = 1.82 Hz, Ar–H), 7.79 (d, 2H, J = 8.24 Hz, Ar–H), 7.62 (d, 1H, J = 8.72 Hz, Ar–H), 7.30 (d, 2H, J = 8.24 Hz, Ar–H), 7.05 (s, 1H, Ar–H), 2.43 (s, 3H, –C3). 13C (100 MHz, CDCl3): δ 161.7, 153.3, 144.5, 140.4, 135.4, 129.7, 126.4, 125.5, 120.2, 118.9, 107.5, 100.6, 21.5. IR (film): 3025, 1600, 1516, 1502, 1345, 915, 874, 816, 730 cm−1. HRMS (EI) calcd for C15H11NO3 [M+] 253.0739; found 253.0731.
:9).1H NMR (400 MHz, CDCl3): δ 8.28 (s, 1H, Ar–H), 7.98 (dd, 1H, J = 8.48 Hz, J = 1.6 Hz, Ar–H), 7.74 (d, 2H, J = 8.24 Hz, Ar–H), 7.51 (d, 1H, J = 8.68 Hz, Ar–H), 7.25 (d, 2H, J = 7.76 Hz, Ar–H), 6.99 (s, 1H, Ar–H), 3.93 (s, 3H, –CO2C3), 2.39 (s, 3H, C3). 13C (100 MHz, CDCl3): δ 167.3, 157.6, 157.3, 139.1, 129.5, 129.4, 127.1, 125.8, 125.2, 125.0, 123.0, 110.9, 100.7, 52.1, 21.4. IR (film): 2952, 1720, 1307, 1244, 1090, 907, 800, 770 cm−1. HRMS (EI) calcd for C17H14O3 [M+] 266.0943; found 266.0949.
:9). 1H NMR (400 MHz, CDCl3): δ 8.32 (d, 1H, J = 1.4 Hz, Ar–H), 8.03 (dd, 1H, J = 8.94 Hz, J = 1.62 Hz, Ar–H), 7.85 (t, 1H, J = 1.6 Hz, Ar–H), 7.74–7.72 (m, 1H, Ar–H), 7.54 (d, 1H, J = 8.68 Hz, Ar–H), 7.41–7.33 (m, 2H, Ar–H), 7.09 (s, 1H, Ar–H), 3.95 (s, 3H, –CO2C3). 13C (100 MHz, CDCl3): δ 167.1, 157.4, 155.7, 135.0, 131.5, 130.1, 128.9, 126.5, 125.5, 125.0, 123.5, 123.1, 111.1, 102.6, 52.1. IR (film): 3108, 1718, 1432, 1276, 912, 821, 762 cm−1. HRMS (EI) calcd for C16H11ClO3 [M+] 286.0397; found 286.0394.
:9). 1H NMR (400 MHz, CDCl3): δ 8.30 (s, 1H, Ar–H), 8.00 (dd, 1H, J = 8.7 Hz, J = 1.82 Hz, Ar–H), 7.85–7.82 (m, 2H, Ar–H), 7.52 (d, 1H, J = 8.72 Hz, Ar–H), 7.15 (t, 2H, J = 8.7 Hz, Ar–H), 6.99 (s, 1H, Ar–H), 3.95 (s, 3H, –CO2C3). 13C (100 MHz, CDCl3): δ 167.2, 163.1 (d, J = 250.05 Hz), 157.3, 156.4, 129.2, 126.9 (d, J = 8.62 Hz), 126.2, 126.1, 125.4, 123.2, 116.0 (d, J = 22.99 Hz), 110.9, 101.2, 52.1. IR (film): 3115, 1719, 1615, 1507, 1301, 1231, 816, 762 cm−1. HRMS (EI) calcd for C16H11FO3 [M+] 270.0692; found 270.0691.
:95). 1H NMR (500 MHz, CDCl3): δ 7.85 (s, 1H, Ar–H), 7.55–7.54 (m, 4H, Ar–H), 7.41–7.36 (m, 3H), 7.34–7.31 (m, 1H, Ar–H), 7.00 (d, 1H, J = 16.2 Hz, Colefin), 6.70 (s, 1H, Ar–H). 13C (125 MHz, CDCl3): δ 157.3, 156.4, 135.9, 132.4, 129.8, 128.9, 128.8, 128.1, 126.9, 125.5, 119.4, 115.3, 111.9, 106.8, 104.1. IR (film): 3029, 2226, 1723, 1590, 1464, 1269, 812, 755, 696 cm−1. HRMS (EI) calcd for C17H11NO [M+] 245.0841; found 245.0842.
olefin), 6.83 (s, 1H, Ar–H). 13C (100 MHz, CDCl3): δ 154.6, 150.3, 136.7, 131.6, 129.3, 128.8, 128.4, 128.0, 126.6, 126.3, 125.1, 124.7, 123.6, 121.1, 120.1, 119.4, 116.6, 106.2. IR (film): 3057, 2925, 1638, 1386, 1084, 951, 815, 746, 685 cm−1. HRMS (EI) calcd for C20H14O [M+] 270.1045; found 270.1042.
olefin), 7.21 (d, 2H, J = 7.76 Hz, Ar–H), 7.05 (d, 1H, J = 16.48 Hz, –Colefin), 6.79 (s, 1H, Ar–H), 2.39 (s, 3H, –C3). 13C (100 MHz, CDCl3): δ 154.8, 150.2, 138.0, 133.9, 131.5, 129.5, 129.3, 128.4, 126.5, 126.3, 125.0, 124.7, 123.5, 121.1, 120.1, 119.4, 115.6, 105.7, 21.3. IR (film): 3052, 2919, 1636, 1519, 1386, 1083, 961, 947, 818, 744 cm−1. HRMS (EI) calcd for C21H16O [M+] 284.1201; found 284.1209.
:95). 1H NMR (400 MHz, CDCl3): δ 8.43 (d, 1H, J = 2.52 Hz, Ar–H), 8.18 (dd, 1H, J = 9.02 Hz, J = 2.4 Hz, Ar–H), 7.53–7.48 (m, 3H, Ar–H), 7.35 (d, 1H, J = 16.48 Hz, –Colefin), 6.93 (d, 2H, J = 8.92 Hz, Ar–H), 6.87 (d, 1H, J = 16.48 Hz, –CHolefin), 6.72 (s, 1H, Ar–H), 3.85 (s, 3H, –OC3). 13C (125 MHz, CDCl3): δ 160.4, 158.8, 157.7, 144.3, 132.4, 129.8, 128.7, 128.5, 120.2, 116.9, 114.4, 113.2, 111.0, 104.1, 55.5. IR (film): 3094, 2924, 1602, 1526, 1510, 1346, 1175, 817 cm−1. HRMS (EI) calcd for C17H13NO4 [M+] 295.0845; found 295.0842.
3). 13C (100 MHz, CDCl3): δ 156.1, 155.4, 153.9, 147.1, 138.6, 138.1, 129.5, 129.4, 128.1, 127.7, 124.8, 124.5, 124.0, 116.1, 114.9, 107.2, 101.3, 97.0, 21.4, 21.3. IR (film): 2922, 2853, 1504, 1244, 1032, 911, 807 cm−1. HRMS (EI) calcd for C24H18O2 [M+] 338.1307; found 338.1309.
:9). 1H NMR (400 MHz, CDCl3): δ 8.14 (s, 1H, Ar–H), 7.88–7.84 (m, 4H, Ar–H), 7.60 (s, 1H, Ar–H), 7.19 (s, 1H, Ar–H), 7.00 (dd, 4H, J = 8.92 Hz, J = 1.6 Hz, Ar–H), 4.02 (s, 3H, –CO2C3), 3.88 (s, 6H, –OC3). 13C (100 MHz, CDCl3): δ 167.3, 160.6, 160.0, 159.0, 156.5, 152.5, 146.7, 126.8, 126.4, 125.1, 123.3, 122.6, 119.2, 116.7, 114.4, 114.3, 109.6, 101.7, 96.1, 55.4, 51.9. IR (KBr): 3011, 2392, 1693, 1647, 1556, 1099, 696 cm−1. HRMS (EI) calcd for C26H20O6 [M+] 428.1260; found 428.1266.
olefin), 2.32 (s, 3H, –C3). 13C (125 MHz, CDCl3): δ 160.8, 152.5, 142.7, 140.1, 129.8, 129.6, 126.0, 125.4, 116.0, 95.4, 21.3. IR (film): 3093, 2923, 1610, 1590, 1517, 1343, 1238, 1111 cm−1. HRMS (EI) calcd for C15H12BrNO3 [M+] 333.0001; found 333.0009.
:95). 1H NMR (400 MHz, CDCl3): δ 7.61 (d, 2H, J = 9.16 Hz, Ar–H), 7.36–7.25 (m, 5H, Ar–H), 7.08 (d, 2H, J = 9.16 Hz, Ar–H), 6.73–6.64 (m, 2H), 6.34 (s, 1H, Colefin). 13C (100 MHz, CDCl3): δ 159.3, 151.9, 135.3, 134.2, 131.6, 128.8, 126.9, 119.7, 118.8, 116.0, 105.8, 99.9. IR (film): 3081, 2226, 1684, 1608, 1502, 1236, 1168 cm−1. HRMS (EI) calcd for C17H12BrNO [M+] 325.0102; found 325.0105.
:98). 1H NMR (400 MHz, CDCl3): δ 8.48 (d, 1H, J = 2.28 Hz, Ar–H), 8.20 (dd, 1H, J = 8.9 Hz, J = 2.52 Hz, Ar–H), 7.77 (d, 2H, J = 8.24 Hz, Ar–H), 7.57 (d, 1H, J = 9.16 Hz, Ar–H), 7.29 (d, 2H, J = 8.24 Hz, Ar–H), 7.06 (s, 1H, Ar–H), 2.42 (s, 3H, –C3). 13C (100 MHz, CDCl3): δ 159.5, 157.5, 144.2, 139.9, 129.7, 126.4, 125.2, 119.8, 117.0, 111.3, 100.8, 21.4. IR (film, cm−1): 2920, 1589, 1519, 1504, 1342, 886, 792, 749. HRMS (ESI) calcd for C15H12NO3 [MH+] 254.0817; found 254.0815.
:95). 1H NMR (400 MHz, CDCl3): δ 7.88 (s, 1H, Ar–H), 7.74 (d, 2H, J = 8.24 Hz, Ar–H), 7.57–7.51 (m, 2H, Ar–H), 7.28–7.24 (m, 2H, Ar–H), 6.98 (s, 1H, Ar–H), 2.40 (s, 3H, –C3). 13C (100 MHz, CDCl3): δ 158.6, 156.3, 139.8, 130.0, 129.6, 127.6, 126.5, 125.5, 125.2, 119.5, 112.1, 106.7, 99.9, 21.4. IR (film): 2923, 2225, 1589, 1463, 1032, 821, 798 cm−1. HRMS (EI) calcd for C16H11NO [M+] 233.0841; found 233.0843.
3). 13C (125 MHz, CDCl3): δ 159.3 (d, J = 236.37 Hz), 157.9, 150.9, 139.0, 130.1 (d, J = 10.8 Hz), 129.5, 127.3, 124.9, 111.6, 111.5, 111.4, 106.1 (d, J = 25.2 Hz), 100.6 (J = 3.6 Hz), 21.4. IR (film): 2961, 1724, 1590, 1461, 1272, 1112, 864, 822, 796 cm−1. HRMS (EI) calcd for C15H11FO [M+] 226.0794; found 226.0799.
3). 13C (125 MHz, CDCl3): δ 157.6, 153.1, 139.1, 130.7, 129.5, 128.3, 127.2, 124.9, 124.0, 120.2, 111.9, 100.0, 21.4. IR (film): 2961, 1725, 1446, 1273, 1035, 824, 808, 796 cm−1. HRMS (EI) calcd for C15H11ClO [M+] 242.0498; found 242.0492.
3). 13C (125 MHz, CDCl3): δ 158.4, 149.0, 139.6, 131.6, 129.5, 128.5, 126.5, 125.1, 123.9, 118.8, 116.9, 100.4, 21.4. IR (film): 2960, 1724, 1575, 1505, 1443, 1277, 1169, 835, 817, 793 cm−1. HRMS (EI) calcd for C15H10Cl2O [M+] 276.0109; found 276.0104.
3). 13C (100 MHz, CDCl3): δ 155.6, 150.1, 138.2, 131.3, 129.5, 128.4, 128.0, 126.3, 124.9, 124.6, 123.5, 121.3, 120.0, 119.5, 101.7, 21.4. IR (film): 3052, 1498, 915, 817, 743 cm−1. HRMS (EI) calcd for C19H14O [M]+ 258.1045; found 258.1046.
3), 2.39 (s, 3H, –C3). 13C (125 MHz, CDCl3): δ 156.2, 153.2, 138.4, 132.2, 129.4, 127.9, 125.2, 124.8, 120.5, 110.5, 100.3, 21.36, 21.33. IR (film): 2962, 1726, 1465, 1289, 823, 800 cm−1. HRMS (EI) calcd for C16H14O [M+] 222.1045; found 222.1043.
3), 2.39 (s, 6H, –C3). 13C (125 MHz, CDCl3): δ 155.3, 154.2, 138.1, 132.4, 129.4, 128.1, 126.6, 124.9, 124.6, 119.6, 117.2, 100.8, 21.3, 19.2, 11.7. IR (film): 3029, 2920, 1726, 1505, 1288, 814 cm−1. HRMS (EI) calcd for C17H16O [M+] 236.1201; found 236.1209.
3), 2.44 (s, 3H, –C3), 2.39 (s, 3H, –C3). 13C (125 MHz, CDCl3): δ 155.0, 154.9, 138.1, 134.2, 130.1, 129.4, 128.0, 126.6, 124.6, 124.5, 108.7, 99.1, 21.7, 21.3, 18.5. IR (film): 3024, 2919, 1727, 1619, 1504, 1294, 1036, 836, 820, 794 cm−1. HRMS (EI) calcd for C17H16O [M+] 236.1201; found 236.1209.
3). 13C (125 MHz, CDCl3): δ 156.1, 154.7, 138.6, 129.4, 129.3, 127.7, 124.8, 123.9, 122.8, 120.7, 111.0, 100.5, 21.4. IR (film): 2932, 1725, 1452, 1270, 801, 749, 737 cm−1. HRMS (EI) calcd for C15H12O [M+] 208.0888; found 208.0880.
:95). 1H NMR (500 MHz, CDCl3): δ 7.91 (s, 1H, Ar–H), 7.87 (d, 2H, J = 7.3 Hz, Ar–H), 7.60–7.54 (m, 2H, Ar–H), 7.48 (t, 2H, J = 7.5 Hz, Ar–H), 7.43–7.40 (m, 1H, Ar–H), 7.05 (s, 1H, Ar–H). 13C (125 MHz, CDCl3): δ 158.3, 156.4, 129.8, 129.5, 129.2, 128.9, 127.8, 125.7, 125.2, 119.4, 112.2, 106.8, 100.7. IR (film): 3093, 2960, 2226, 1723, 1465, 1288, 896, 825, 747, 766 cm−1. HRMS (ESI) calcd for C15H10NO [MH+] 220.0762; found 220.0769.
:95). 1H NMR (500 MHz, CDCl3): δ 7.90 (s, 1H, Ar–H), 7.78 (d, 2H, J = 8.6 Hz, Ar–H), 7.58–7.54 (m, 2H, Ar–H), 7.44 (d, 2H, J = 8.05 Hz, Ar–H), 7.02 (s, 1H, Ar–H). 13C (125 MHz, CDCl3): δ 157.1, 156.4, 135.4, 129.7, 129.2, 128.1, 127.7, 126.4, 125.8, 119.3, 112.3, 107.0, 101.1. IR (film): 2960, 2225, 1723, 1486, 1461, 1269, 1090, 810, 793 cm−1. HRMS (EI) calcd for C15H8ClNO [M+] 253.0294; found 253.0290.
:98). 1H NMR (500 MHz, CDCl3): δ 8.51 (s, 1H, Ar–H), 8.23 (d, 1H, J = 9.2 Hz, Ar–H), 7.81 (d, 2H, J = 8 Hz, Ar–H), 7.59 (d, 1H, J = 9.2 Hz, Ar–H), 7.47 (d, 2H, J = 8 Hz, Ar–H), 7.12 (s, 1H, Ar–H). 13C (125 MHz, CDCl3): δ 158.1, 157.6, 144.4, 135.6, 129.5, 129.3, 127.7, 126.5, 120.3, 117.4, 111.5, 102.0. IR (film): 3099, 2961, 1722, 1588, 1520, 1486, 1348, 1268, 1090, 793 cm−1. HRMS (EI) calcd for C14H8ClNO3 [M+] 273.0193; found 273.0197.
3), 2.40 (s, 3H, –C3). 13C (125 MHz, CDCl3): δ 154.4, 153.9, 133.8, 133.0, 129.3, 128.9, 126.3, 125.9, 125.1, 119.8, 117.5, 102.0, 19.3, 11.7. IR (film): 3037, 2921, 1725, 1481, 1090, 814, 750 cm−1. HRMS (EI) calcd for C16H13ClO [M+] 256.0655; found 256.0650.
3). 13C (100 MHz, CDCl3): δ 159.7, 155.4, 149.9, 131.2, 128.4, 126.2, 126.1, 125.0, 124.7, 123.6, 123.4, 121.3, 119.9, 119.4, 114.3, 100.8, 55.3. IR (film): 2926, 1500, 1252, 1032, 814, 748 cm−1. HRMS (EI) calcd for C19H14O2 [M]+ 274.0994; found 274.0985.
:98). 1H NMR (400 MHz, CDCl3): δ 7.77 (d, 2H, J = 9.16 Hz, Ar–H), 7.50 (d, 1H, J = 1.84 Hz, Ar–H), 7.40 (d, 1H, J = 8.68 Hz, Ar–H), 7.19 (dd, 1H, J = 8.68 Hz, J = 1.84 Hz, Ar–H), 6.98 (d, 2H, J = 8.68 Hz, Ar–H), 6.81 (s, 1H, Ar–H), 3.86 (s, 3H, –OC3). 13C (100 MHz, CDCl3): δ 160.3, 157.5, 153.0, 130.8, 128.3, 126.5, 123.8, 122.7, 120.0, 114.3, 111.9, 99.1, 55.3. IR (film): 2964, 1724, 1505, 1271, 1040, 832, 796 cm−1. HRMS (EI) calcd for C15H11ClO2 [M+] 258.0448; found 258.0445.
:98). 1H NMR (500 MHz, CDCl3): δ 7.79 (d, 2H, J = 6.85 Hz, Ar–H), 7.39 (s, 1H, Ar–H), 7.25–7.23 (m, 1H, Ar–H), 6.97 (d, 2H, J = 7.45 Hz, Ar–H), 6.81 (s, 1H, Ar–H), 3.86 (s, 3H, –OC3). 13C (125 MHz, CDCl3): δ 160.6, 158.3, 149.0, 131.8, 128.5, 126.8, 123.7, 122.1, 118.7, 116.9, 114.3, 99.5, 55.4. IR (film): 2967, 1720, 1610, 1505, 1445, 1255, 824, 792 cm−1. HRMS (EI) calcd for C15H10Cl2O2 [M+] 292.0058; found 292.0050.
3). 13C (100 MHz, CDCl3): δ 157.0, 150.5, 139.1, 130.9, 129.5, 127.1, 125.1, 124.1, 123.7, 119.2, 116.5, 100.9, 21.4. IR (film): 3033, 1589, 1471, 1193, 1034, 905, 805, 734 cm−1. HRMS (EI) calcd for C15H11ClO [M+] 242.0498; found 242.0497.
3). 13C (100 MHz, CDCl3): δ 157.0, 154.8, 138.9, 129.5, 128.0, 127.3, 124.9, 123.6, 121.1, 111.6, 100.2, 21.4. IR (film): 3092, 1582, 1503, 1423, 1030, 925, 816 cm−1. HRMS (EI) calcd for C15H11ClO [M+] 242.0498; found 242.0495.
:9).1H NMR (400 MHz, CDCl3): δ 8.40 (s, 1H, Ar–H), 8.17 (dd, 1H, J = 8.72 Hz, J = 1.82 Hz, Ar–H), 7.79 (d, 2H, J = 8.24 Hz, Ar–H), 7.62 (d, 1H, J = 8.72 Hz, Ar–H), 7.30 (d, 2H, J = 8.24 Hz, Ar–H), 7.05 (s, 1H, Ar–H), 2.43 (s, 3H, –C3). 13C (100 MHz, CDCl3): δ 161.7, 153.3, 144.5, 140.4, 135.4, 129.7, 126.4, 125.5, 120.2, 118.9, 107.5, 100.6, 21.5. IR (film): 3025, 1600, 1516, 1502, 1345, 915, 874, 816, 730 cm−1. HRMS (EI) calcd for C15H11NO3 [M+] 253.0739; found 253.0731.
:9).1H NMR (400 MHz, CDCl3): δ 8.28 (s, 1H, Ar–H), 7.98 (dd, 1H, J = 8.48 Hz, J = 1.6 Hz, Ar–H), 7.74 (d, 2H, J = 8.24 Hz, Ar–H), 7.51 (d, 1H, J = 8.68 Hz, Ar–H), 7.25 (d, 2H, J = 7.76 Hz, Ar–H), 6.99 (s, 1H, Ar–H), 3.93 (s, 3H, –CO2C3), 2.39 (s, 3H, C3). 13C (100 MHz, CDCl3): δ 167.3, 157.6, 157.3, 139.1, 129.5, 129.4, 127.1, 125.8, 125.2, 125.0, 123.0, 110.9, 100.7, 52.1, 21.4. IR (film): 2952, 1720, 1307, 1244, 1090, 907, 800, 770 cm−1. HRMS (EI) calcd for C17H14O3 [M+] 266.0943; found 266.0949.
:9). 1H NMR (400 MHz, CDCl3): δ 8.32 (d, 1H, J = 1.4 Hz, Ar–H), 8.03 (dd, 1H, J = 8.94 Hz, J = 1.62 Hz, Ar–H), 7.85 (t, 1H, J = 1.6 Hz, Ar–H), 7.74–7.72 (m, 1H, Ar–H), 7.54 (d, 1H, J = 8.68 Hz, Ar–H), 7.41–7.33 (m, 2H, Ar–H), 7.09 (s, 1H, Ar–H), 3.95 (s, 3H, –CO2C3). 13C (100 MHz, CDCl3): δ 167.1, 157.4, 155.7, 135.0, 131.5, 130.1, 128.9, 126.5, 125.5, 125.0, 123.5, 123.1, 111.1, 102.6, 52.1. IR (film): 3108, 1718, 1432, 1276, 912, 821, 762 cm−1. HRMS (EI) calcd for C16H11ClO3 [M+] 286.0397; found 286.0394.
:9). 1H NMR (400 MHz, CDCl3): δ 8.30 (s, 1H, Ar–H), 8.00 (dd, 1H, J = 8.7 Hz, J = 1.82 Hz, Ar–H), 7.85–7.82 (m, 2H, Ar–H), 7.52 (d, 1H, J = 8.72 Hz, Ar–H), 7.15 (t, 2H, J = 8.7 Hz, Ar–H), 6.99 (s, 1H, Ar–H), 3.95 (s, 3H, –CO2C3). 13C (100 MHz, CDCl3): δ 167.2, 163.1 (d, J = 250.05 Hz), 157.3, 156.4, 129.2, 126.9 (d, J = 8.62 Hz), 126.2, 126.1, 125.4, 123.2, 116.0 (d, J = 22.99 Hz), 110.9, 101.2, 52.1. IR (film): 3115, 1719, 1615, 1507, 1301, 1231, 816, 762 cm−1. HRMS (EI) calcd for C16H11FO3 [M+] 270.0692; found 270.0691.
:95). 1H NMR (500 MHz, CDCl3): δ 7.85 (s, 1H, Ar–H), 7.55–7.54 (m, 4H, Ar–H), 7.41–7.36 (m, 3H), 7.34–7.31 (m, 1H, Ar–H), 7.00 (d, 1H, J = 16.2 Hz, Colefin), 6.70 (s, 1H, Ar–H). 13C (125 MHz, CDCl3): δ 157.3, 156.4, 135.9, 132.4, 129.8, 128.9, 128.8, 128.1, 126.9, 125.5, 119.4, 115.3, 111.9, 106.8, 104.1. IR (film): 3029, 2226, 1723, 1590, 1464, 1269, 812, 755, 696 cm−1. HRMS (EI) calcd for C17H11NO [M+] 245.0841; found 245.0842.
olefin), 6.83 (s, 1H, Ar–H). 13C (100 MHz, CDCl3): δ 154.6, 150.3, 136.7, 131.6, 129.3, 128.8, 128.4, 128.0, 126.6, 126.3, 125.1, 124.7, 123.6, 121.1, 120.1, 119.4, 116.6, 106.2. IR (film): 3057, 2925, 1638, 1386, 1084, 951, 815, 746, 685 cm−1. HRMS (EI) calcd for C20H14O [M+] 270.1045; found 270.1042.
olefin), 7.21 (d, 2H, J = 7.76 Hz, Ar–H), 7.05 (d, 1H, J = 16.48 Hz, –Colefin), 6.79 (s, 1H, Ar–H), 2.39 (s, 3H, –C3). 13C (100 MHz, CDCl3): δ 154.8, 150.2, 138.0, 133.9, 131.5, 129.5, 129.3, 128.4, 126.5, 126.3, 125.0, 124.7, 123.5, 121.1, 120.1, 119.4, 115.6, 105.7, 21.3. IR (film): 3052, 2919, 1636, 1519, 1386, 1083, 961, 947, 818, 744 cm−1. HRMS (EI) calcd for C21H16O [M+] 284.1201; found 284.1209.
:95). 1H NMR (400 MHz, CDCl3): δ 8.43 (d, 1H, J = 2.52 Hz, Ar–H), 8.18 (dd, 1H, J = 9.02 Hz, J = 2.4 Hz, Ar–H), 7.53–7.48 (m, 3H, Ar–H), 7.35 (d, 1H, J = 16.48 Hz, –Colefin), 6.93 (d, 2H, J = 8.92 Hz, Ar–H), 6.87 (d, 1H, J = 16.48 Hz, –CHolefin), 6.72 (s, 1H, Ar–H), 3.85 (s, 3H, –OC3). 13C (125 MHz, CDCl3): δ 160.4, 158.8, 157.7, 144.3, 132.4, 129.8, 128.7, 128.5, 120.2, 116.9, 114.4, 113.2, 111.0, 104.1, 55.5. IR (film): 3094, 2924, 1602, 1526, 1510, 1346, 1175, 817 cm−1. HRMS (EI) calcd for C17H13NO4 [M+] 295.0845; found 295.0842.
3). 13C (100 MHz, CDCl3): δ 156.1, 155.4, 153.9, 147.1, 138.6, 138.1, 129.5, 129.4, 128.1, 127.7, 124.8, 124.5, 124.0, 116.1, 114.9, 107.2, 101.3, 97.0, 21.4, 21.3. IR (film): 2922, 2853, 1504, 1244, 1032, 911, 807 cm−1. HRMS (EI) calcd for C24H18O2 [M+] 338.1307; found 338.1309.
:9). 1H NMR (400 MHz, CDCl3): δ 8.14 (s, 1H, Ar–H), 7.88–7.84 (m, 4H, Ar–H), 7.60 (s, 1H, Ar–H), 7.19 (s, 1H, Ar–H), 7.00 (dd, 4H, J = 8.92 Hz, J = 1.6 Hz, Ar–H), 4.02 (s, 3H, –CO2C3), 3.88 (s, 6H, –OC3). 13C (100 MHz, CDCl3): δ 167.3, 160.6, 160.0, 159.0, 156.5, 152.5, 146.7, 126.8, 126.4, 125.1, 123.3, 122.6, 119.2, 116.7, 114.4, 114.3, 109.6, 101.7, 96.1, 55.4, 51.9. IR (KBr): 3011, 2392, 1693, 1647, 1556, 1099, 696 cm−1. HRMS (EI) calcd for C26H20O6 [M+] 428.1260; found 428.1266.
Acknowledgements
We thank Council of Scientific and Industrial Research (CSIR), New Delhi for funding this work (No. 02(0091)/12/EMR-II). P.D. acknowledges CSIR for the research fellowship.Notes and references
- G. Chelucci, Chem. Rev., 2012, 112, 1344–1462 CrossRef CAS PubMed.
- (a) M. L. N. Rao, D. N. Jadhav and P. Dasgupta, Org. Lett., 2010, 12, 2048–2051 CrossRef CAS PubMed; (b) M. L. N. Rao, D. N. Jadhav and P. Dasgupta, Eur. J. Org. Chem., 2013, 781–788 CrossRef CAS.
- (a) S. Thielges, E. Meddah, P. Bisseret and J. Eustache, Tetrahedron Lett., 2004, 45, 907–910 CrossRef CAS; (b) J. Liu, W. Chen, Y. Ji and L. Wang, Adv. Synth. Catal., 2012, 354, 1585–1592 CrossRef CAS; (c) G. A. Kraus and V. Gupta, Org. Lett., 2010, 12, 5278–5280 CrossRef CAS PubMed.
- (a) S. Rizzo, A. Tarozzi, M. Bartolini, G. D. Costa, A. Bisi, S. Gobbi, F. Belluti, A. Ligresti, M. Allara, J.-P. Monti, V. Andrisano, V. D. Marzo, P. Hrelia and A. Rampa, Eur. J. Med. Chem., 2012, 58, 519–532 CrossRef CAS PubMed; (b) Y.-X. Tan, C. Liu and R.-Y. Chen, Phytochem. Lett., 2012, 5, 419–422 CrossRef CAS; (c) X.-F. Duan, G. Shen and Z.-B. Zhang, Synthesis, 2010, 2547–2552 CrossRef CAS; (d) S.-Y. Lin, C.-L. Chen and Y.-J. Lee, J. Org. Chem., 2003, 68, 2968–2971 CrossRef CAS PubMed; (e) J. Zhang, L. Li, Y. Wang, W. Wang, J. Xue and Y. Li, Org. Lett., 2012, 14, 4528–4530 CrossRef CAS PubMed; (f) H.-D. Choi, P.-J. Seo, B.-W. Son and B.-W. Kang, Arch. Pharmacal Res., 2003, 26, 985–989 CrossRef CAS; (g) Q. Li, J. Min, Y.-H. Anh, J. Namm, E. M. Kim, R. Lui, H. Y. Kim, Y. Ji, H. Wu, T. Wisniewski and Y.-T. Chang, ChemBioChem, 2007, 8, 1679–1687 CrossRef CAS PubMed; (h) M. Matsumoto, S. Dobashi and K. Kondo, Bull. Chem. Soc. Jpn., 1977, 50, 3026–3028 CrossRef CAS; (i) H. Tsuji, L. Ilies and E. Nakamura, Synlett, 2014, 2099–2110 CrossRef CAS; (j) G. A. Kraus and V. Gupta, Tetrahedron Lett., 2009, 50, 7180–7183 CrossRef CAS.
- (a) E. Ruijter, R. Scheffelaar and R. V. A. Orru, Angew. Chem., Int. Ed., 2011, 50, 6234–6246 CrossRef CAS PubMed; (b) N. T. Patil, V. S. Shinde and B. Gajula, Org. Biomol. Chem., 2012, 10, 211–224 RSC; (c) L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS PubMed; (d) L. F. Tietze and U. Beifuss, Angew. Chem., Int. Ed., 1993, 32, 131–163 CrossRef; (e) B. M. Trost, Angew. Chem., Int. Ed., 1995, 34, 259–281 CrossRef CAS.
- F. Ramirez, N. B. Desai and N. Mckelvie, J. Am. Chem. Soc., 1962, 84, 1745–1747 CrossRef CAS.
- H. Hopf and B. Witulski, in Modern Acetylene Chemistry, ed. P. J. Stang and F. Diederich, VCH, Weinheim, Germany, 1995, ch. 2, pp. 33–66 Search PubMed.
- A. Wagner, M. P. Heitz and C. Mioskowski, Tetrahedron Lett., 1990, 31, 3141–3144 CrossRef CAS.
- T. Nishikawa, S. Shibuya, S. Hosokawa and M. Isobe, Synlett, 1994, 485–486 CrossRef CAS.
- S. Wang, P. Li, L. Yu and L. Wang, Org. Lett., 2011, 13, 5968–5971 CrossRef CAS PubMed.
- G. A. Molander and C. R. Bernardi, J. Org. Chem., 2002, 67, 8424–8429 CrossRef CAS PubMed.
- (a) P. Colbon, J. H. Barnard, M. Purdie, K. Mulholland, I. Kozhevnikov and J. Xiao, Adv. Synth. Catal., 2012, 354, 1395–1400 CrossRef CAS; (b) T. T. B. Nguyen, T. Lomberget, N. C. Tran, E. Colomb, L. Nachtergaele, S. Thoret, J. Dubois, J. Guillaume, R. Abdayem, M. Haftek and R. Barret, Bioorg. Med. Chem. Lett., 2012, 22, 7227–7231 CrossRef CAS PubMed; (c) M. Jacubert, O. Provot, J.-F. Peyrat, A. Hamez, J.-D. Brion and M. Alami, Tetrahedron, 2010, 66, 3775–3787 CrossRef CAS; (d) L. M. Geary and P. G. Hultin, Org. Lett., 2009, 11, 5478–5481 CrossRef CAS PubMed; (e) M. Nakamura, L. Ilies, S. Otsubo and E. Nakamura, Org. Lett., 2006, 8, 2803–2805 CrossRef CAS PubMed; (f) A. R. Katritzky, C. N. Fali and J. Li, J. Org. Chem., 1997, 62, 8205–8209 CrossRef CAS PubMed; (g) A. Kasahara, T. Izumi and T. Ogihara, J. Heterocycl. Chem., 1989, 26, 597–599 CrossRef CAS.
- L. M. Geary and P. G. Hultin, Eur. J. Org. Chem., 2010, 5563–5573 CrossRef CAS.
- M. L. N. Rao, P. Dasgupta and V. N. Murty, RSC Adv., 2015, 5, 24834–24845 RSC.
- W. Chen, P. Li, T. Miao, L.-G. Meng and L. Wang, Org. Biomol. Chem., 2013, 11, 420–424 CAS.
- E. A. Jaseer, D. J. C. Prasad and G. Sekar, Tetrahedron, 2010, 66, 2077–2082 CrossRef CAS.
- S. Nandi, S. Kumar, H. Ila and H. Junjappa, ARKIVOC, 2010, 22–31 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1406195. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra13213d |
| This journal is © The Royal Society of Chemistry 2015 |