
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCalcium-modified hierarchically porous aluminosilicate geopolymer as a highly efficient regenerable catalyst for biodiesel production†
Sudhanshu
Sharma
a,
Dinesh
Medpelli
b,
Shaojiang
Chen
b and
Dong-Kyun
Seo
*b
aDepartment of Chemistry, Indian Institute of Technology, Gandhinagar, VGEC campus, Chandkheda, Ahmedabad, Gujarat, India 382424
bDepartment of Chemistry and Biochemistry, Arizona State University, Tempe, Arizona 85287-1604, USA. E-mail: dseo@asu.edu
First published on 27th July 2015
Abstract
A new class of highly active solid base catalysts for biodiesel production was developed by creating hierarchically porous aluminosilicate geopolymer with affordable precursors and modifying the material with varying amounts of calcium. For the catalysts containing ≥8 wt% Ca, almost 100% conversion has been achieved in one hour under refluxing conditions with methanol solvent, and the high catalytic activity was preserved for multiple regeneration cycles. Temperature-programed desorption studies of CO2 indicate that the new base catalyst has three different types of base sites on its surface whose strengths are intermediate between MgO and CaO. The detailed powder X-ray diffraction (PXRD) and X-ray photoelectron spectroscopic (XPS) studies show that the calcium ions were incorporated into the aluminosilicate network of the geopolymer structure, resulting in a very strong ionicity of the calcium and thus the strong basicity of the catalysts. Little presence of CaCO3 in the catalysts was indicated from the thermogravimetric analysis (TGA), XPS and Fourier transform infrared spectroscopy (FT-IR) studies, which may contribute to the observed high catalytic activity and regenerability. The results indicate that new geopolymer-based catalysts can be developed for cost-effective biodiesel production.
Introduction
Biodiesel is an important biofuel which can be produced from a variety of biogenic oils through transesterification.1–3 For efficient conversion, the reaction is catalyzed by acid or base catalysts which are chosen judiciously, depending upon the nature of the biogenic oil.4 Base catalysts are usually preferred because of their significantly higher activity than acid catalysts.5 Alkali hydroxides such as KOH and NaOH are some of the most commonly used base homogeneous catalysts for biodiesel synthesis.6 Although homogeneous catalysts have advantages due to the molecular level interactions which give rise to a high activity, heterogeneous catalysts (solid catalysts) are often preferred because of their ease of separation and reusability.3,5A number of different solid base catalysts have been studied for biodiesel synthesis including alkaline earth metal oxides and rare earth oxides.2–4,7 Among many different catalytic compositions CaO is particularly of high importance because of its high basicity, low toxicity, abundance and cost effectiveness.8–10 However, several reports have indicated that calcium ions leach out from the catalyst during the transesterification because of the appreciable solubility of CaO in methanol and hence that the biodiesel product needs to be purified with decalcifying agents at an added cost for a high purity of the final products.4,9,10 Moreover, active sites on CaO are easily poisoned by atmospheric CO2 due to formation of carbonate and also by water through hydration, which requires calcination at 700 °C in order to reactivate the catalyst at the expense of surface area.11 It has been demonstrated that such regenerability problems can be solved by supporting CaO on silica (SBA-15) or CeO2.12,13 However, the high cost or rarity of the support materials can be a disadvantage in practice.
Recently, zeolites have been studied as an alternative support materials partly due to their commercial availability at an affordable cost. By using CaO nanoparticles supported on NaX zeolite (Si/Al = 1.3; 5–25 wt% CaO), the transesterification reaction could be carried out at the reflux temperature of methanol (65 °C) under an atmospheric pressure, with a good efficiency, which has been attributed to the fact that the amount of basic sites increases by the introduction of alkali earth oxides.14 NaY has also been shown as a promising support with a certain degree of water and acid resistance.15 While this approach is promising, catalytic deactivation was observed due to formation of carbonate.14–16 In addition, it is noted that the commercial zeolites are in the form of typically micron-sized particles and thus they have relatively low surface area for the CaO particles to deposit. Given the large molecular weights of triglycercides (average MW > 800 g mol−1),17 triglycerides can hardly access the zeolite micropores (pore aperture ≤ 8 Å),18,19 and hence the innate high surface area from the microporosity is not operative for biodiesel production. Introduction of larger pore sizes in the zeolites, or more generally in alkali aluminosilicates with similar chemical structures and compositions, may enhance the efficacy of the materials and thus realize the potential of this strategy.
Geopolymers, amorphous alkali-activated aluminosilicates, are a promising engineered material which has been increasingly studied as a more environment-friendly alternative to Portland cement in construction due to their large-scale availability, excellent thermal stability and superior mechanical properties.20–22 Chemically, they are similar to low-silica zeolites in that their amorphous chemical structure consists of a three-dimensional network of AlO4 and SiO4 tetrahedra connected by oxygen corners with Si/Al ratios typically from 1 to 3. Microstructural analysis of geopolymer has indicated that the geopolymer is an innate nanomaterial with a xerogel-like structure made up of highly fused nanoparticles (10–30 nm in diameter)23 and that the core of the nanoparticles is more likely zeolitic.24 The dense nature of the geopolymer microstructure narrows the pore channels in the xerogel-like structure, preventing effective molecular flow, as evidenced from the ill-shaped hysteresis loops in gas sorption isotherms.25
Nevertheless, new advanced applications of geopolymer, including catalysis26–28 and drug delivery,29 are emerging, by taking advantage of the innate nanostructure of geopolymer. In a recent study by Sazama et al. for example, modified geopolymers have been shown to be a promising heterogeneous catalyst for selective reduction of nitrogen oxides by ammonia and the total oxidation of volatile hydrocarbons.26 Catalytic sites were generated by modifying Na-based or K/Ca-based geopolymer through an ion exchange process using aqueous solutions of transition metal ions including Fe3+, Co2+, Cu2+ and Pt2+. In similar synthetic approaches in which conventional geopolymer is employed as a starting material, photocatalytic activities have been realized in Ni2+-ion exchanged geopolymer and TiO2-containing geopolymer. Therefore, geopolymer system may be promising to discover new effective catalysts and the advance in this area may be expedited by a more deliberate control of the nanostructures of geopolymer with high surface area and high porosity.
Herein, we demonstrate for the first time that a high-surface area high-porosity geopolymer can be an excellent transesterification catalyst for biodiesel production when it is modified with calcium. It was achieved by employing a new type of porous geopolymer that exhibits a hierarchical pore structure with macropores and mesopores, reported in our recent work.20 Details of the preparation, characterization and catalyst test results are given for biodiesel synthesis using soybean oil. Effect of calcium loading, reaction time and reusability test are given to indicate the effectiveness of the new catalyst. X-ray photoelectron spectroscopic studies before and after the transesterification reaction were also carried out to elucidate the nature of calcium at the catalyst surface.
Experimental
Synthesis of catalysts
The synthesis of porous geopolymer was carried out by following the procedure detailed in our recent publication.20 The composition with the nominal K![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al:Si ratios of 2:1:2 was chosen in our studies as it resulted in the highest pore volume and surface area. Namely, 6.1 g of KOH pellets (≥85%, Sigma Aldrich) were first dissolved in deionized water in a polypropylene cup. 3.33 g of fumed silica (Cabot, CABOSIL® EH-5) was added to this solution and the mixture was stirred with mechanical mixer (IKA RW20 Digital Mixer) for 30 min at 800 rpm to give a clear solution. 6.17 g of metakaolinite was slowly added into the mixture solution to form a homogenous fluidic liquid called as geopolymer resin. The metakaolinite was prepared in advance by calcining kaolinite (Al2Si2O7·H2O, Alfa Aesar) at 750 °C for 10 h. Canola oil (The J.M. Smucker Company, Crisco®) was added to the geopolymer resin at a 1:1 oil-to-water volume ratio and mixed for additional 15 min to give a homogeneous but viscous blend. This blend was cured at 60 °C in a laboratory oven for 24 h, giving a solid monolith which was ground to a coarse powder. The powder was washed repeatedly with butanol to remove the organic components and further washed multiple times with deionized water to remove the excess alkali. The product, porous geopolymer, was dried at 110 °C overnight and stored for the next step.
:Al:Si ratios of 2:1:2 was chosen in our studies as it resulted in the highest pore volume and surface area. Namely, 6.1 g of KOH pellets (≥85%, Sigma Aldrich) were first dissolved in deionized water in a polypropylene cup. 3.33 g of fumed silica (Cabot, CABOSIL® EH-5) was added to this solution and the mixture was stirred with mechanical mixer (IKA RW20 Digital Mixer) for 30 min at 800 rpm to give a clear solution. 6.17 g of metakaolinite was slowly added into the mixture solution to form a homogenous fluidic liquid called as geopolymer resin. The metakaolinite was prepared in advance by calcining kaolinite (Al2Si2O7·H2O, Alfa Aesar) at 750 °C for 10 h. Canola oil (The J.M. Smucker Company, Crisco®) was added to the geopolymer resin at a 1:1 oil-to-water volume ratio and mixed for additional 15 min to give a homogeneous but viscous blend. This blend was cured at 60 °C in a laboratory oven for 24 h, giving a solid monolith which was ground to a coarse powder. The powder was washed repeatedly with butanol to remove the organic components and further washed multiple times with deionized water to remove the excess alkali. The product, porous geopolymer, was dried at 110 °C overnight and stored for the next step.
Incipient wetness technique30 was used to load calcium ions in the porous geopolymer. A known concentration of Ca(NO3)2 solution in methanol was prepared and added dropwise to a finely ground porous geopolymer (500 mg) until the sample became nearly wet homogeneously. The sample was then dried in a laboratory oven at 80 °C and calcined in a muffle furnace at 550 °C for 8 h. In total, seven solutions with different Ca(NO3)2 concentrations from 1 to 7 wt% were used to change the calcium loading. Table 1 shows the sample names used throughout this report, along with the selected results from the characterization described in the next section.
| Catalyst | Amount of Caa (wt%) | Pore properties | Yieldd (%) | ||
|---|---|---|---|---|---|
| Surface area (m2 g−1) | Pore volumeb (cm3 g−1) | Pore widthc (nm) | |||
| a Determined by ICP-OES. b From the pores with width no larger than 150 nm in the BJH desorption pore distribution. c 4(BJH desorption pore volume)/(BET surface area). d Determined by 1H NMR spectroscopy. | |||||
| GEO | 0 | 108 | 0.50 | 18 | 23 |
| CaGEO-1 | 2.2 | 94 | 0.45 | 19 | 80 |
| CaGEO-2 | 5.3 | 79 | 0.41 | 19 | 93 |
| CaGEO-3 | 7.5 | 76 | 0.38 | 19 | 100 |
| CaGEO-4 | 8.8 | 75 | 0.39 | 19 | 100 |
| CaGEO-5 | 10 | 65 | 0.34 | 20 | 100 |
| CaGEO-6 | 12 | 62 | 0.32 | 20 | 100 |
| CaGEO-7 | 18 | 55 | 0.30 | 21 | 100 |
Characterization of catalysts
Powder X-ray diffraction (PXRD) patterns of the finely ground samples were collected using a Siemens D5000 diffractometer with CuKα radiation (λ = 1.5407 Å). N2 sorption isotherms were obtained with a Micrometrics ASAP 2020 volumetric adsorption analyzer at 77 K. Samples were degassed at room temperature for 10 h under vacuum until a residual pressure of ≤10 μm Hg was reached. Specific surface areas were estimated using Brunauer–Emmett–Teller (BET) equation, in the relative pressure (p/po) range from 0.06 to 0.2.31 Pore volumes were calculated from the amount of nitrogen adsorbed at p/po = 0.99. Pore size distributions were obtained by applying the Barrett–Joyner–Halenda (BJH) model to the desorption branch of the N2 sorption isotherms, assuming a cylindrical pore shape.32Samples for scanning electron microscopy (SEM) were prepared by placing small pieces of the products (approximate cubes of few millimeters in length) on a SEM stub using a copper conducting tape. Samples were then gold coated for 150 s and were studied using SEM-XL30 Environmental FEG (FEI) microscope operating at 10 kV. Thermogravimetric analysis (TGA) studies were carried out using a Mettler Toledo TGA/DSC 1 instrument equipped with GC 200 gas controller. Samples were placed in 70 μL alumina crucibles and were analyzed by heating from 25 to 800 °C at a heating rate of 10 °C min−1 under an air flow at 50 mL min−1. Fourier transform infrared spectroscopy (FT-IR) was recorded with Thermo Scientific Nicolet 6700 FT-IR Spectrometer. The spectra were collected on finely ground samples in an attenuated total reflectance (ATR) mode using a liquid nitrogen cooled MCT detector.
The calcium loadings of the samples were determined by using Thermo Scientific iCAP 6300 inductively coupled plasma-optical emission spectrometer (ICP-OES). Prior to the analysis, solid samples were acid-digested using a CEM MARS 6 microwave reaction system in repeated heating steps at 180 °C for 30 min with sequential addition of required reagents. Specifically, 20–30 mg of catalysts were heated in the reactor first with 3 mL of concentrated HCl solution (34–37 wt%, ACS), and second with a mixture of 3 mL concentrated HNO3 (67–70 wt%, ACS) and 0.5 mL of HF solution (48–51 wt%, ACS). The digests were later quenched with 5 mL of 4.5 wt% H3BO3 solution aided by the microwave reactor.
X-ray photoelectron spectroscopic studies (XPS) were carried out using a VG ESCALAB 220I-XL XPS system (Thermo VG Scientific Ltd., UK) with a monochromated Al Kα radiation (1486.6 eV, line width 0.7 eV) in order to examine the nature of calcium ions in the catalyst samples before and after the transesterification reaction. Binding energies were corrected with respect to the C(1s) peak at 284.5 eV. The pressure in the analyzing chamber was kept at the level of 10−9 Torr while recording the spectra and the spectrometer had the energy resolution of 0.4 eV.
In order to investigate the surface basicity of the catalysts, temperature-programmed desorption (TPD) was performed with CO2 using a flow system connected to a Flame Ionization Detector (FID).33 50 mg of geopolymer catalyst sample (sieve size 80–250 μm) was placed in a quartz tube (6 mm OD, 4 mm ID and 25 cm in length) and heated in a N2 gas flow from room temperature to 700 °C at a heating rate of 10 °C min−1 with a flow rate of 30 mL min−1. After reaching the final temperature, the gas flow was switched to CO2 at the same flow rate and the sample was cooled down to room temperature. The CO2 gas flow was then stopped and the sample was flushed in nitrogen gas for 30 minutes at room temperature in order to remove any weakly adsorbed gas molecules including CO2. Thereafter, the sample was heated in presence of N2 gas with the same flow rate from room temperature to 700 °C at 10 °C min−1 while the amount of desorbed CO2 was monitored with the FID.
Catalytic transesterification of soybean oil
The activity of the catalysts were examined through transesterification of soybean oil in a refluxing condition of methanol at an ambient pressure.34 All experiments were carried out in a 250 mL round-bottom flask equipped with a magnetic stirrer and a water-cooled condenser. 100 mg of the catalyst and 6 g of anhydrous methanol (Mallinckrodt Chemicals, 99.8% min, ACS) were added to the flask and this mixture was stirred with a magnetic bar at 1000 rpm for 15 min to disperse the catalyst. The temperature was then raised to reach a refluxing condition (boiling point of methanol = 67 °C). 1 g of soybean oil (Spectrum Laboratory Products, Inc.) was added to the reaction mixture and the mixture was kept stirring at 300 rpm under the same reaction condition for a given period of time. The amount of the catalyst was 1.4% of the total weight of the reaction mixture solution.After the reaction (2 h of reaction time, unless mentioned otherwise), the mixture was cooled and hexanes (BDH, 98.5% min. C6 isomers; 60% min n-C6) were added to the reaction mixture and mixed homogeneously. The whole mixture was transferred into a 20 mL centrifuge tube. After centrifugation for 15 min at 5000 rpm, two liquid layers were visible; the upper layer of hexanes containing biodiesel and possibly unreacted oil and the bottom layer having methanol and glycerol. After filtering the catalyst out, the top layer was transferred into a beaker and placed in a laboratory oven at 70 °C to evaporate the hexanes. The content of the biodiesel in the residue was analyzed by employing a 1H NMR spectroscopy with a 400 MHz Varian NMR spectrometer, following the method described in literature.35 Briefly, the transesterification yield was calculated from the ratio of the integrated areas between the singlet peak of methoxy protons in methyl esters at ∼3.6 ppm and the triplet peaks of methylene protons in both triglycerides and methyl esters at ∼2.2 ppm, respectively:

Hot filtration test was conducted to confirm that the new catalysts were indeed heterogeneous.36–38 In this test, the same reaction procedure was followed except that after 10 min of the reaction, the catalyst was filtered off from the hot reaction mixture with a porous alumina filter and the reaction in the filtrate was then continued under the same refluxing condition for additional 50 min.
Results and discussion
The calcium loadings estimated from the ICP-OES results are given in Table 1. Approximately 2 to 18 wt% calcium loadings has been achieved using Ca(NO3)2 solutions of different concentrations. The values increase from CaGEO-1 to CaGEO-7, as we expect from the increased calcium concentration employed for the preparation of the samples. PXRD patterns of geopolymers catalysts (GEO to CaGEO-7) show an amorphous nature with a broad hump centered at 2θ from 28 to 30° (Fig. 1). No Bragg reflection peaks related to CaO were observed in any of the PXRD patterns of the samples. Presence of amorphous CaO is less likely because the calcination temperature (500 °C) was high enough to provide a noticeable crystallinity to CaO. The hump is located around 30° for the unmodified geopolymer, GEO, and the hump gradually shifts to a lower angle down to 28°, indicating that the increasing incorporation of calcium results in a slight but continuous increment in interatomic distances. This is in opposite to the observed decrease in interatomic distances for the K-geopolymers with partially ion-exchanged Ca ions.39 One reason for this puzzling behaviour can be that in contrast to the “exchange” of cations in the previous studies, Ca ions may be incorporated in the geopolymer matrix together with additional oxide anions in the final calcined structure and thus increase the average atomic distances. | ||
| Fig. 1 Powder X-ray diffraction patterns of unmodified geopolymer (GEO) and calcium-modified geopolymers (CaGEO-1 to -7) from bottom to top. See Table 1 for the details of the samples. The small sharp peak at 25.3° is from an anatase TiO2 impurity (PDF# 00-021-1272) in the metakaolin. | ||
XPS studies on the catalysts have been carried out and a representative high-resolution Ca(2p) core level spectrum is shown for CaGEO-4 in Fig. 2. Binding energies of Ca2p3/2 and Ca2p1/2 are 349.4 and 352.8 eV, respectively and these values are significantly higher than those of a number of different calcium containing compounds,40 indicating a strong ionicity of the Ca2+ ions, or equivalently a strong ionic character in the Ca–O bonding in the catalysts. For example, the Ca2p3/2 energy for the catalysts is 2–3 eV higher than that of CaO41,42 and thus confirms further that the Ca2+ ions do not exist in the form of CaO in the catalysts. Indeed, the large value is in the same range with the energy values for Ca(NO3)2 (348.7 eV)43 and for Ca-ion exchange zeolite Y (348.6 eV).44 The high ionicity is expected for Ca2+ ions surrounded by a covalent aluminosilicate network of geopolymer.
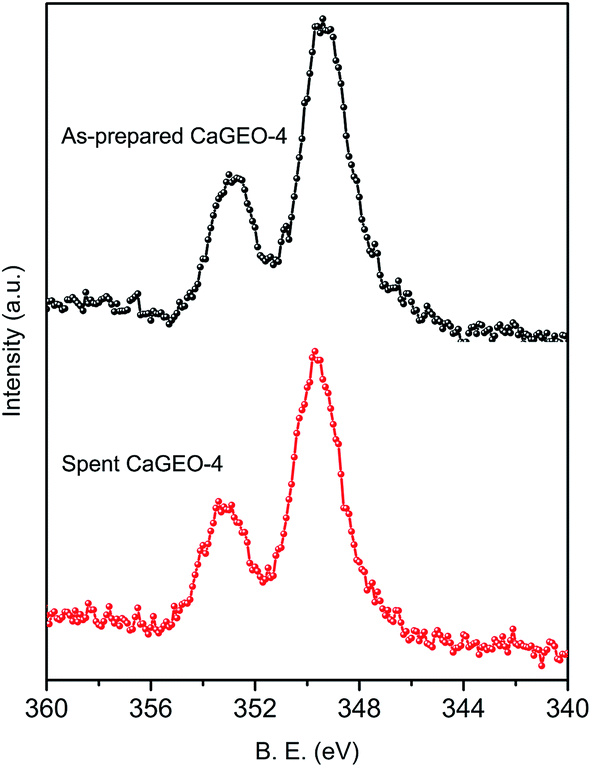 | ||
| Fig. 2 Ca(2p) core level spectra of as-prepared and spent catalyst CaGEO-4. Binding energies are corrected using C(1s) at 284.5 eV. | ||
Fig. 3a shows the N2 sorption isotherms of the samples. They all exhibit type-IV isotherms with H2-type hysteresis loop, indicating the presence of mesopores. The corresponding BJH curves point to relatively narrow pore size distributions with the maximum around 20 nm (Fig. 3b). A small upward shift of the maximum position is noted as the Ca loading increases, which corroborates the trend in the average pore size (from 18 to 21 nm) observed in Table 1. Meanwhile, the mesopore volume and BET surface area decrease continuously, which may indicate that the pores are enlarged by more effective sintering and consolidation of the geopolymer nanoparticles. Fig. 4 shows scanning electron microscope (SEM) images of GEO and CaGEO-4 as a representative example. The materials exhibits discrete spherical pores whose diameters range from about 5 to 40 μm in Fig. 4a and b. A closer look in Fig. 4c and d reveals that the pore wall separating the spherical pores has a finer structure throughout the matrix, which are consistent with our previous results that the hierarchically porous geopolymer products were found to exhibit a mesoporous matrix made up of rather leisurely connected amorphous aluminosilicate nanoparticles and that large spherical macropores were scattered over throughout the mesoporous matrix.20 It is noted that the fine structures observed for both GEO and Ca-GEO4 are similar, if not indistinguishable, as we might expect from the similar pore characteristics found from the N2 gas sorption analysis.
 | ||
| Fig. 3 N2 gas isotherms (a), and BJH pore distribution curves (b) of unmodified geopolymer (GEO) and calcium-modified geopolymers (CaGEO-1 to -7) from top to bottom. The grey dashed lines indicate the zero starting points for the vertical axis. See Table 1 for the details of the samples. | ||
 | ||
| Fig. 4 SEM images of GEO in (a) and (c) (scale bar = 20 and 2 μm, respectively) and of CaGEO-4 in (b) and (d) (scale bar = 20 and 2 μm, respectively). | ||
The TPD profile of CO2 desorbed from CaGEO-4 in Fig. S1† shows a main peak at around 350 °C with two shoulders at 300 and 425 °C. Since the temperature of desorption represents the strength of the adsorbate–adsorbent interaction, Fig. S1† indicates presence of more than one type of basic sites which may be weak, moderate and strong. This surface site heterogeneity is not inconsistent with the complex chemical nature of the amorphous geopolymer surface exchanged with Ca2+ ions. All the peak positions are lower than the value reported for CaO (490 °C)45 but higher than the value for the most basic site on MgO (270 °C).46,47 This indicates that the basicity of CaGEO-4 is between CaO and MgO, although a caution needs to be paid in comparing literature data since TPD profiles depend on experimental conditions such as the amount of the sample, flow rate of the carrier gas, the rate of temperature rise in addition to the nature of basic sites.33 Integration of the total peak areas indicates that 0.77 mmol g−1 (i.e., ∼9 μmol m−2) of CO2 was desorbed from CaGEO-4. The amount is relatively high among base catalysts.33 These results substantiate that the Ca-geopolymer material has surface Ca sites with a considerable strength of basicity, unique from CaO and other alkaline earth metal oxides.
Fig. 5a shows the biodiesel yield as a function of calcium loading for the catalysts after 2 h of the refluxing reaction, which was obtained from 1H NMR spectra of the products. A representative 1H NMR spectra of the products is given in Fig. S1 (ESI†) together with that of a biodiesel standard (VHG Labs, 100% Biodiesel, Lot No. 107-0084), and both exhibit the identical features. It is noted that the unmodified geopolymer (GEO) itself can catalyze the transesterification of the soybean oil but at a low yield (∼23%). This yield did not improve even with increasing the reaction time from 2 to 24 h. CaGEO-1 (2.2 wt% Ca) substantially increased the yield to 80% and the yield reaches 100% for CaGEO-3 (7.5 wt% Ca) and the catalysts with a higher calcium loading (Fig. 5a).
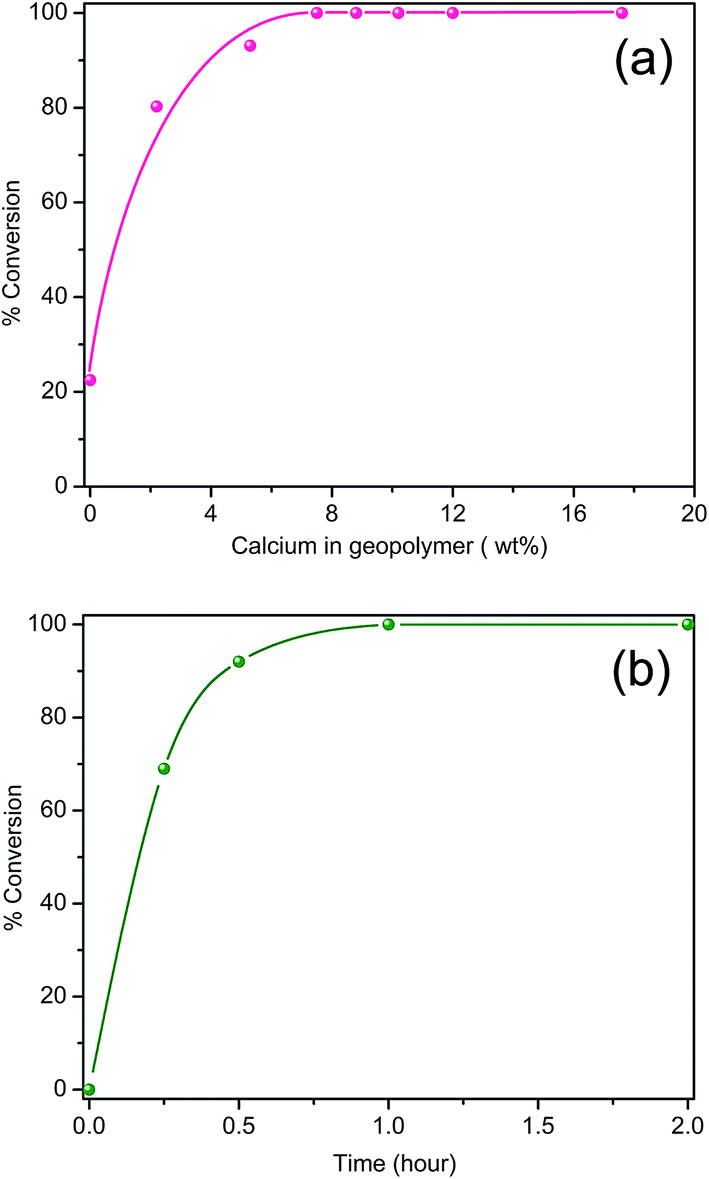 | ||
| Fig. 5 Soybean oil-to-biodiesel conversion yield (%) for calcium modified geopolymer catalyst as a function of calcium amount (a), and as a function of reaction time (b) under the same refluxing condition at 67 °C for CaGEO-4. | ||
The reaction yield was then monitored for CaGEO-4 as a function of the reaction time under the same refluxing reaction condition. As observed in Fig. 5b, the transesterification reaction is fast at the beginning with a 70% conversion within 15 min. The reaction slows down after 30 min, but it reaches 100% eventually in 1 h. These results fair well with the solid base catalysts reported in the literature, in that previous works have reported typical yields no greater than 95% after 1–3 h of methanol reflux reactions when CaO itself or CaO supported on zeolites.3,8,11,14,15,48 For example, CaO catalysts showed a <95% conversion of sunflower or soybean oil in 1.5 to 3 h under the same refluxing condition.49,50 In other works, CaO nanoparticles supported on NaX zeolite (Si/Al = 1.3; 5–25 wt% CaO), resulted in up to ∼94% yield from sunflower oil,14 while more recently, a biodiesel yield of 95% from soybean oil was achieved by using the CaO/NaY catalyst (Si/Al = 3.24), with a rather high catalyst loading (30 wt%).15
The recyclability of the catalysts was examined by repetitive cycles of a catalytic refluxing reaction and subsequent catalyst regeneration by calcination at 500 °C for two h. Fig. 6 shows the conversion yield up to 8 cycles for CaGEO-4 and the results indicate that the catalyst maintains its catalytic activity up to about 5 cycles and the gradual decrease in the activity is apparent afterwards. It is not clear what is responsible to the observed decrease in the catalytic activity. Fig. 7 shows the PXRD patterns of CaGEO-4 before and after the cycled processes and they do not show any apparent differences, indicating insignificant structural changes, if any, after the processes. This is consistent with the Ca(2p) core level spectra of the spent catalyst CaGEO-4 in Fig. 2 which did not show noticeable change after the repeated reactions. A small blue shift by about 0.3 eV for both Ca2p3/2 and Ca2p1/2 peaks is noticed in the high resolution XPS spectrum after the reactions, but the shift is too small to conclude a definite change in the chemical environment around calcium ions. One may speculate that the calcium ions in the catalysts do not experience appreciable chemical changes during the repeated use but the cause for the deactivation after multiple cycles is still unknown.
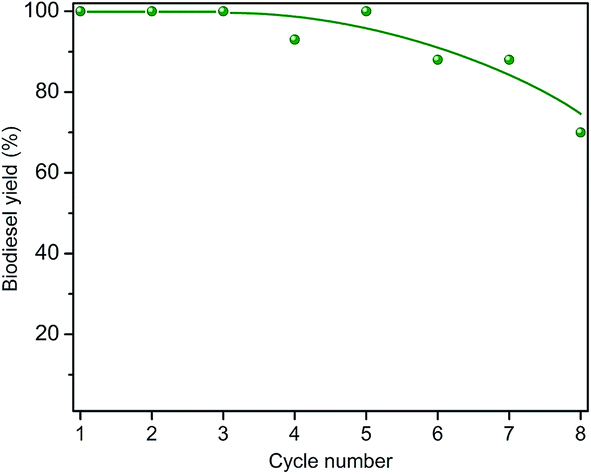 | ||
| Fig. 6 Soybean oil-to-biodiesel conversion yield (%) versus number of catalytic cycles for the CaGEO-4 catalyst under the same refluxing condition at 67 °C. After each cycle, the catalyst was washed and calcined. | ||
 | ||
| Fig. 7 Powder X-ray diffraction patterns of as-prepared and spent catalyst CaGEO-4. | ||
In order to check whether the Ca-geopolymer catalyzes the reaction heterogeneously or not, CaGEO-4 was subjected to a hot filtration test in which the catalyst was removed from the reaction mixture at the end of 10 min and the reaction was continued further without the catalyst for a total of 1 h period.36–38 The product yield from 1H NMR spectrum was 32%, which is in agreement with what we would expect after a 10 min reaction period from Fig. 5b. This indicates that the reaction did not proceed upon the removal of the catalyst and thus confirms that the Ca-geopolymer is indeed a heterogeneous catalyst with no leaching of active Ca species. Upon the assumption that aluminosilicate does not dissolve in methanol, the elemental analysis based on the XPS spectra of the as-synthezed and spent CaGEO-4 indicates that about 4 at% of Ca was lost from the surface of the catalyst during the 8 cycles of the repeated reactions. This implies that only 0.5 at% of the Ca ions were released on average to the reaction medium and the value corresponds to ∼70 ppm of Ca concentration in each batch of the reaction mixture solution. This estimated Ca concentration is too low to be a major catalytic agent for the biesel production. It also indicates that leaching of calcium ions from our catalyst is insignificant and that the product is relatively free from metal ions.
TGA thermograms of the as-prepared and spent CaGEO-4 (after 8 cycles) samples are shown in Fig. 8a, along with that of the unmodified geopolymer (GEO). Overall, GEO shows a significant weight loss (9.3 wt%) up to 500 °C, at first due to the evaporation of free or physisorbed water in the pores below 200 °C and later because of the loss of chemisorbed water.51 Such weight losses are less prominent for the as-prepared CaGEO-4 (6.3 wt%) and even less after 8 cycles of catalysis reactions and regenerations (3.3 wt%). When comparing the as-prepared CaGEO-4 with GEO, the smaller adsorbed water amount is understandable because of the lower surface area that the former has, as shown in Table 1. Indeed, the ratio of the weight loss (9.3/6.3 = 1.5) is very close to the surface area ratio (108/75 = 1.4). Unfortunately, the same argument cannot be made concretely for the spent CaGEO-4, because its surface area value is not available due to the gradual sample loss over the 8 cycles. Nevertheless, it is expected for this sample to have the least surface area and hence exhibit the minimum amount of surface water due to the fact that it underwent 8 high temperature regeneration steps, one each after each of the eight catalytic cycles.
 | ||
| Fig. 8 TGA thermograms (a) and FT-IR spectra (b) of GEO, as-prepared CaGEO-4, and spent CaGEO-4 after 8 catalytic cycles. | ||
Unlike GEO, both as-prepared and spent CaGEO-4 samples exhibit an additional weight loss step from about 650 to 800 °C (1.2 and 0.9 wt%, respectively). Considering the chemical compositions of the samples and the temperature range, the weight loss can be accounted for by a possible presence of CaCO3 which can be formed by adsorption of atmospheric CO2 and subsequent carbonation of the strong basic sites on calcium species within the pores. The decomposition of nanosized CaCO3 into CO2(g) and CaO occurs at temperatures as low as 650 °C, as reported in the literature.52 The presence of CaCO3 is supported by the FT-IR spectra of the three samples in Fig. 8b, in which only the CaGEO-4 samples exhibit the characteristic peak of carbonates around 1450 cm−1.53 Considering the appreciable amount of Ca (8.8 wt%) in the catalyst, the significantly small weight losses (1.2 and 0.9%) of the samples indicate that the most of the Ca species are carbonate-free and that CaCO3 may have formed only at the surface of the pores of the catalyst. This is consistent with our previous conclusion from the PXRD and XPS studies that Ca ions are incorporated into the amorphous chemical structure of the aluminosilicate geopolymer. The observed low extent of carbonation, probably at the surface, allows the catalyst to be regenerated relatively easily without hampering the porosity and high surface area, rendering the catalyst highly regenerable.
Concluding remarks
We have successfully prepared a new class of high-surface area solid base catalysts based on modification of hierarchically porous aluminosilicate geopolymer. The catalysts have shown a significantly high catalytic activity. Namely, with only 7.5 wt% Ca loading in the structure, the catalyst could achieve 100% conversion of soybean oil to a biodiesel within 1 h under an ambient refluxing condition with methanol as a solvent. The catalysts are recyclable without losing the original activity for about 5 cycles after which the conversion yield decreases gradually. The detailed powder X-ray diffraction and X-ray photoelectron spectroscopic studies indicate that the calcium ions must be incorporated into the geopolymer structure, resulting in a very strong ionicity of the calcium and thus strong basicity for the catalysts. It is concluded that the new geopolymer-based catalysts can be an excellent choice for cost-effective biodiesel production owing to their high catalytic activity and good regenerability and yet futher research is necessary for realization of its potential.References
- A. Demirbas, Prog. Energy Combust. Sci., 2005, 31, 466–487 CrossRef CAS PubMed.
- F. Maa and M. A. Hanna, Bioresour. Technol., 1999, 70, 1–15 CrossRef.
- A. A. Refaat, Int. J. Environ. Sci. Technol., 2011, 8, 203–221 CrossRef CAS.
- Y. C. Sharma, B. Singh and J. Korstad, Fuel, 2011, 90, 1309–1324 CrossRef CAS PubMed.
- Z. Helwani, M. R. Othman, N. Aziz, J. Kim and W. J. N. Fernando, Appl. Catal., A, 2009, 363, 1–10 CrossRef CAS PubMed.
- J. M. Dias, M. C. M. Alvim-Ferraz and M. F. Almeida, Fuel, 2008, 87, 3572–3578 CrossRef CAS PubMed.
- D. Singh, R. Bhoi, A. Ganesh and S. Mahajani, Energy Fuels, 2014, 28, 2743–2753 CrossRef CAS.
- X. Liu, H. He, Y. Wang, S. Zhu and X. Piao, Fuel, 2008, 87, 216–221 CrossRef CAS PubMed.
- P.-L. Boey, G. P. Maniam and S. A. Hamid, Chem. Eng. J., 2011, 168, 15–22 CrossRef CAS PubMed.
- S. Yan, C. DiMaggio, S. Mohan, M. Kim, S. Salley and K. Y. S. Ng, Top. Catal., 2010, 53, 721–736 CrossRef CAS.
- M. L. Granados, M. D. Z. Poves, D. M. Alonso, R. Mariscal, F. C. Galisteo, R. Moreno-Tost, J. Santamaría and J. L. G. Fierro, Appl. Catal., B, 2007, 73, 317–326 CrossRef CAS PubMed.
- M. C. G. Albuquerque, I. Jiménez-Urbistondo, J. Santamaría-González, J. M. Mérida-Robles, R. Moreno-Tost, E. Rodríguez-Castellón, A. Jiménez-López, D. C. S. Azevedo, C. L. Cavalcante Jr and P. Maireles-Torres, Appl. Catal., A, 2008, 334, 35–43 CrossRef CAS PubMed.
- W. Thitsartarn and S. Kawi, Green Chem., 2011, 13, 3423–3430 RSC.
- S. Luz Martinez, R. Romero, J. C. Lopez, A. Romero, V. Sanchez Mendieta and R. Natividad, Ind. Eng. Chem. Res., 2011, 50, 2665–2670 CrossRef CAS.
- H. Wu, J. Zhang, Q. Wei, J. Zheng and J. Zhang, Fuel Process. Technol., 2013, 109, 13–18 CrossRef CAS PubMed.
- P. Kovacheva, A. Predoeva, K. Arishtirova and S. Vassilev, Appl. Catal., A, 2002, 223, 121–128 CrossRef CAS.
- Industrial Crops And Uses, ed. B. P. Singh, CAB International, Wallingford, England, 2010 Search PubMed.
- Zeolites in industrial separation and catalysis, ed. S. Kulprathipanja, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010 Search PubMed.
- D. R. Taylor, C. B. Ungermann and Z. Demidowicz, J. Am. Oil Chem. Soc., 1984, 61, 1372–1379 CrossRef CAS.
- D. Medpelli, J.-M. Seo and D.-K. Seo, J. Am. Ceram. Soc., 2013, 7, 70–73 Search PubMed.
- J. Davidovits, J. Therm. Anal., 1991, 37, 1633–1656 CrossRef CAS.
- P. Duxson, A. Fernandez-Jimenez, J. L. Provis, G. C. Lukey, A. Palomo and J. S. J. v. D. Duxson, J. Mater. Sci., 2007, 42, 2917–2933 CrossRef CAS.
- W. M. Kriven, J. L. Bell and M. Gordon, Ceram. Trans., 2003, 153, 227–250 CAS.
- J. L. Provis, G. C. Lukey and J. S. J. van Deventer, Chem. Mater., 2005, 17, 3075–3085 CrossRef CAS.
- P. Duxson, J. L. Provis, G. C. Lukey, S. W. Mallicoat, W. M. Kriven and J. S. J. van Deventer, Colloids Surf., A, 2005, 269, 47–58 CrossRef CAS PubMed.
- P. Sazama, O. Bortnovsky, J. Dedecek, Z. Tvaruzkova and Z. Sobalík, Catal. Today, 2011, 164, 92–99 CrossRef CAS PubMed.
- J. R. Gasca-Tirado, A. Manzano-Ramirez, P. A. Vazquez-Landaverde, E. I. Herrera-Diaz, M. E. Rodriguez-Ugarte, J. C. Rubio-Avalos, V. Amigo-Borras and M. Chavez-Paez, Mater. Lett., 2014, 134, 222–224 CrossRef CAS PubMed.
- Y. J. Zhang, L. C. Liu, Y. Xu, Y. C. Wang and D. L. Xu, J. Hazard. Mater., 2012, 209–210, 146–150 CrossRef CAS PubMed.
- E. Jaemstorp, M. Stromme and G. Frenning, J. Pharm. Sci., 2011, 100, 4338–4348 CrossRef CAS PubMed.
- Handbook of Heterogeneous Catalysis, Wiley-VCH, Weinheim, Germany, 2nd edn, 2008 Search PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- Y. Ono and H. Hattori, Solid Base Catalysis, Springer-Verlag, Berlin, Heidelberg, 2011 Search PubMed.
- C. Ngamcharussrivichai, W. Wiwatnimit and S. Wangnoi, J. Mol. Catal. A: Chem., 2007, 276, 24–33 CrossRef CAS PubMed.
- G. Gelbard, O. Bres, R. M. Vargas, F. Vielfaure and U. F. Schuchardt, J. Am. Oil Chem. Soc., 1995, 72, 1239–1241 CrossRef CAS.
- M. Guidotti and B. Lázaro, in Deactivation and Regeneration of Zeolite Catalysts, Imperial College Press, 2011, vol. 9, pp. 303–334 Search PubMed.
- I. W. C. E. Arends and R. A. Sheldon, Appl. Catal., A, 2001, 212, 175–187 CrossRef CAS.
- H. Gruber-Woelfler, P. F. Radaschitz, P. W. Feenstra, W. Haas and J. G. Khinast, J. Catal., 2012, 286, 30–40 CrossRef CAS PubMed.
- G. M. Canfield, J. Eichler, K. Griffith and J. D. Hearn, J. Mater. Sci., 2014, 49, 5922–5933 CrossRef CAS.
- NIST, National Institute of Standards and Technology, Gaithersburg, Editon edn, 2012.
- H. van Doveren and J. A. T. H. Verhoeven, J. Electron Spectrosc. Relat. Phenom., 1980, 21, 265–273 CrossRef CAS.
- H. Seyama and M. Soma, J. Chem. Soc., Faraday Trans., 1984, 80, 237–248 RSC.
- A. B. Christie, J. Lee, I. Sutherland and J. M. Walls, Appl. Surf. Sci., 1983, 15, 224–237 CrossRef CAS.
- T. L. Barr, Appl. Surf. Sci., 1983, 15, 1–35 CrossRef CAS.
- G. Zhang, H. Hattori and K. Tanabe, Appl. Catal., 1988, 36, 189–197 CrossRef CAS.
- V. K. Diez, C. R. Apesteguia and J. I. Di Cosimo, J. Catal., 2003, 215, 220–233 CrossRef CAS.
- J. I. Di Cosimo, V. K. Diez, C. Ferretti and C. R. Apesteguia, in Catalysis: Volume 26, The Royal Society of Chemistry, 2014, vol. 26, pp. 1–28 Search PubMed.
- A. K. Endalew, Y. Kiros and R. Zanzi, Biomass Bioenergy, 2011, 35, 3787–3809 CrossRef CAS PubMed.
- M. L. Granados, M. D. Z. Poves, D. M. Alonso, R. Mariscal, F. C. Galisteo, R. Moreno-Tost, J. Santamaria and J. L. G. Fierro, Appl. Catal., B, 2007, 73, 317–326 CrossRef CAS PubMed.
- X. Liu, H. He, Y. Wang, S. Zhu and X. Piao, Fuel, 2007, 87, 216–221 CrossRef PubMed.
- J. L. Bell, P. Sarin, P. E. Driemeyer, R. P. Haggerty, P. J. Chupas and W. M. Kriven, J. Mater. Chem., 2008, 18, 5974–5981 RSC.
- S. Wang, Z. Cui, X. Xia and Y. Xue, Phys. B, 2014, 454, 175–178 CrossRef CAS PubMed.
- H. Böke, S. Akkurt, S. Özdemir, E. H. Göktürk and E. N. Caner Saltik, Mater. Lett., 2004, 58, 723–726 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra01823d |
| This journal is © The Royal Society of Chemistry 2015 |