DOI:
10.1039/C5RA12705J
(Paper)
RSC Adv., 2015,
5, 87019-87029
Synthesis of new U-shaped azobenzene liquid crystals for photoswitching properties†
Received
30th June 2015
, Accepted 8th October 2015
First published on 8th October 2015
Abstract
A new series of liquid crystalline compounds comprising a U-shaped unit as central core incorporating azobenzene in the side arms and having terminal alkene functional groups, is synthesized and characterized by differential scanning calorimetry (DSC), polarized-light optical microscopy (POM), X-ray diffraction analysis and UV-vis spectroscopy. In the U-shaped series, all compounds showed stable enantiotropic monolayer SmA phases independent on the chain length and chain parity. These U-shaped molecules exhibit strong photoisomerisation behaviour in solutions and in solid state. The photoswitching properties of compounds showed trans to cis isomerization in about 15 seconds, whereas reverse process required much longer times ranging from 235–380 min in solutions. In case of solid film of 4a, E–Z photoisomerization takes around 4 s and the reverse transformation to original Z–E state takes about 74 min. The kinetic study using UV light irradiation shows that all compounds (4a–e) behave first order rate law throughout the relaxation time in solution. The effect of alkyl chain length for trans to cis is negligible whereas, cis to trans is substantial on the odd–even chain length. The photoisomerization study with variable light intensities shows that the photosaturation and thermal back relaxation times are decreased when intensity of the irradiated light is increased. Compounds did not degrade with light illumination at 10 mW cm−2. The reversible isomerization did not significantly decay after multiple cycles indicating that the photo-responsive properties are stable and repeatable. Thus, the photoswitching behaviour of these materials may be suitably exploited in the field of optical data storage device and in molecular switches for suitable switching times.
1. Introduction
Anisometric molecules (mesogens), either rod-shaped or disc-shaped, are used to design and synthesize conventional thermotropic calamitic or discotic liquid crystals.1–4 Generally liquid crystals (LCs) produced with rod-like molecules display nematic and/or smectic mesophases whereas LCs with flat disc-shaped molecules display nematic and/or columnar mesophases.1,5 Comparatively a new class of LCs whose molecular shape is distorted away from the classical rod or disc shape is designated as non-conventional LCs, for instance oligomeric LCs, bent-core or banana LCs, polycatenars, and dendrimers.1,5–8 Certainly, the non-classical molecular architectures may exhibit mesophases even though their molecular geometry deviate significantly from the classical rod or disc-shapes.1
Vorlander and Apel9,10 realized the synthesis of the first U-shaped or bent-shaped molecules as early as in 1929 using the disubstituted benzene ring substituted either at 1,2 or 1,3-positions which deviates significantly from classical rod-shaped molecules. Yelamaggad et al.1 reported bent core V-shaped mesogens consisting of salicylaldimine mesogenic segments derived from 1,2-substitution of benzene ring and other reported compounds having 1,2-substitution of benzene ring, also termed as bent core V-shaped molecules.10–12 Literature survey reveal that some works are reported12–15 for the U-shaped molecular architectures using the 1,2-substituted phenylene compounds which are very relevant to this work. Therefore, we wish to designate the term U-shaped molecules in this paper. The U-shaped molecules exhibit mesophases which are analogous to classical calamitic LCs, whereas banana-shaped mesogens exhibit new type of smectic phases, which are not identical to the phases formed by calamitics.1 U-shaped molecule, viz. 1,2-phenylene bis[4-(ethoxyphenylazoxy) benzoate] was first reported by Vorlander and Apel10 and this compound exhibit a nematic phase which was identified by Pelzl et al.11 Three series of compounds were reported by Kuboshita et al.12 and these compounds showed nematic, smectic A, and smectic B phases. The bent-core compounds are found as fused twins13 or U-shaped molecules14 are also reported. A homologous series of U-shaped dimeric liquid crystals in which two mesogenic groups were linked to catechol was reported by Yoshizawa and co-workers.15 The odd–even effect is found on the phase sequences in which the even members favour smectic C phases, whereas the odd members favour smectic A phases; however, both members exhibits the nematic phase.15 Catechol based U-shaped dimeric liquid crystals incorporating to 3 to 6 methylene units with terminal aliphatic chain lengths varying from 1 to 12 units were prepared by Attard et al.16 In the two homologous series, compounds with odd number of methylene units form nematic and smectic phases as a function of terminal chain length whereas compounds with even number of methylene units are smectogenic. X-ray diffraction studies confirm that these smectic phases are composed of molecules arranged in bilayers.16 Recent study show that two rod-shaped azobenzene moieties, each carrying a short electron withdrawing acetyl group at the terminals when attached to a catechol unit via methylene spacers exhibits nematic and smectic A phases irrespective of chain length.17 Matsuzaki and Matsunaga18 reported different series of V-shaped molecules and these compounds were also shows the N, SmA and SmB phases. A partial bilayer structure was proposed based on X-ray diffraction studies for the SmA phase. Kato et al.19 reveal the nematic phase of V-shaped molecules which is formed from the hydrogen bonding between phthalic acid and stilbazole. Tanaka and Yoshizawa20 reported a homologous series of catechol derivatives having 2,3-difluoro-1,4-diphenylbenzene including one of the biphenyl derivatives. The phase transition behavior of each U-shaped compound doped with a chiral dopant reveal that some compounds show the blue phases and investigation performed in terms of helical twisting power, elasticity and molecular biaxiality, which enable to informed about the effects of the U-shapes on stabilization of blue phases.20 Some V-shaped tetradentate ligands were prepared by condensing 1,2-phenylenediamine with benzaldehyde compounds which exhibit mesomorphic behavior.21
Alongside with the design and synthesis of liquid crystals molecules, a field of research namely the photoinduced phenomenon such as the incident light promoting the molecular ordering/disordering of the liquid-crystalline system gained a lot of credence.22–25 In photonics, where light can be controlled as a stimulus, is offering as the future technology for high-speed information processing. In such systems the reversible photoinduced shape transformation of the molecules containing the photochromic azobenzene groups take place.26–28 Thus, liquid crystals with an azo-linkage have received attention due to their unique photoswitchable properties induced by light.29–33 Upon absorption of UV light (∼365 nm), the energetically more stable E configuration (trans) converts to the Z configuration (cis). The reverse transformation of the Z isomer into the E isomer can be shifted by irradiation with visible light (in the range of 400 to 500 nm) the process is known as thermal back relaxation which occurs in the dark or thermally or photochemically with visible light.34–37
So far, 1,2-phenylene bis[4-{[4-(alkyloxy)phenyl]diazenyl} benzoate] moieties have not been employed to perceive such U-shaped dimeric mesogens. In this paper, we have synthesized a series of new molecules in which photoswitchable azo moieties are attached to the 1,2-phenylene as a core via ether linkage. These compounds exhibit smectic A phase irrespective of chain length and parity. Further, we report some E/Z isomerization studied on the unconventional azobenzene compounds for possible applications in the optical data storage and molecular switches.
2. Experimental
2.1 Synthesis of intermediate compounds
All intermediate compounds 1, 2a–e and 3a–e were synthesized according to our earlier paper.22–24
2.2 1,2-Phenylene bis[4-{[4-(but-3-en-1-yloxy)phenyl]diazenyl} benzoate] (4a)
Compound 3a (0.1060 g, 0.358 mmol), catechol (0.0196 g, 0.179 mmol), 50 ml of dry dichloromethane, DMAP (4.8 mg, 0.04 mmol) and DCC (0.0820 g, 0.40 mmol) were stirred for 48 h. Esterification and purifications were carried out according to our previous paper.22,25 Yield of 4a: 0.0410 g, 33%. IR, νmax/cm−1 3070 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 2929 (CH2), 2857 (CH2), 1736 (CO, ester), 1639 (CC, vinyl), 1605, 1496 (CC, aromatic), 1254, 1130, 1062 (C–O), 830 (C–H). δH(500 MHz; CDCl3; Me4Si) 8.24 (d, 4H, J = 8.6 Hz, Ph), 7.92 (d, 4H, J = 8.9 Hz, Ph), 7.85 (d, 4H, J = 8.5 Hz, Ph), 7.47 (m, 2H, Ph), 7.42 (m, 2H, Ph), 7.02 (d, 4H, J = 8.9 Hz, Ph), 5.91–5.86 (m, 2H), 5.05 (dd, 2H, J = 13.6 Hz), 4.96 (dd, 2H, J = 9.7 Hz), 4.05 (t, 4H, J = 6.5 Hz, OCH2–), 2.10–2.04 (m, 4H, –CH2–). δC(125 MHz; CDCl3; Me4Si) 26.85, 67.69, 109.12, 110.78, 121.75, 122.81, 124.29, 125.34, 126.12, 131.55, 136.39, 146.78, 152.56, 156.47, 162.65, 164.12.
CH2), 2929 (CH2), 2857 (CH2), 1736 (CO, ester), 1639 (CC, vinyl), 1605, 1496 (CC, aromatic), 1254, 1130, 1062 (C–O), 830 (C–H). δH(500 MHz; CDCl3; Me4Si) 8.24 (d, 4H, J = 8.6 Hz, Ph), 7.92 (d, 4H, J = 8.9 Hz, Ph), 7.85 (d, 4H, J = 8.5 Hz, Ph), 7.47 (m, 2H, Ph), 7.42 (m, 2H, Ph), 7.02 (d, 4H, J = 8.9 Hz, Ph), 5.91–5.86 (m, 2H), 5.05 (dd, 2H, J = 13.6 Hz), 4.96 (dd, 2H, J = 9.7 Hz), 4.05 (t, 4H, J = 6.5 Hz, OCH2–), 2.10–2.04 (m, 4H, –CH2–). δC(125 MHz; CDCl3; Me4Si) 26.85, 67.69, 109.12, 110.78, 121.75, 122.81, 124.29, 125.34, 126.12, 131.55, 136.39, 146.78, 152.56, 156.47, 162.65, 164.12.
2.3 1,2-Phenylene bis[4-{[4-(pent-4-en-1-yloxy)phenyl]diazenyl} benzoate] (4b)
Compound 4b was synthesis from 3b according to the method described for 4a. Yield of 4b: 0.1081 g, 35%. IR, νmax/cm−1 3072 (CH2), 2942 (CH2), 2883 (CH2), 1737 (νCO, ester), 1638 (CC, vinyl), 1600, 1499 (CC, aromatic), 1259, 1137, 1062 (C–O), 855 (C–H). δH(500 MHz; CDCl3; Me4Si) 8.22 (d, 4H, J = 8.7 Hz, Ph), 7.91 (d, 4H, J = 9.0 Hz, Ph), 7.86 (d, 4H, J = 8.7 Hz, Ph), 7.46 (m, 2H, Ph), 7.41 (m, 2H, Ph), 7.01 (d, 4H, J = 9.0 Hz, Ph), 5.90–5.87 (m, 2H), 5.10 (dd, 2H, J = 15.5 Hz), 5.04 (dd, 2H, J = 10.1 Hz), 4.06 (t, 4H, J = 6.5 Hz, OCH2–), 2.27–2.24 (m, 4H, –CH2–), 1.94–1.91 (m, 4H, –CH2–). δC(125 MHz; CDCl3; Me4Si) 27.55, 30.14, 67.66, 109.85, 110.98, 121.55, 122.32, 124.21, 125.65, 126.04, 131.47, 136.57, 146.71, 152.26, 156.11, 162.44, 164.55.
2.4 1,2-Phenylene bis[4-{[4-(hex-5-en-1-yloxy)phenyl]diazenyl} benzoate] (4c)
Compound 4c was synthesis from 3c according to the method described for 4a. Yield of 4c: 0.0630 g, 37%. IR, νmax/cm−1 3071 (CH2), 2924 (CH2), 2854 (CH2), 1730 (CO, ester), 1640 (CC, vinyl), 1604, 1497 (CC, aromatic), 1258, 1141, 1081 (C–O), 800 (C–H). δH(500 MHz; CDCl3; Me4Si) 8.24 (d, 4H, J = 8.5 Hz, Ph), 7.92 (d, 4H, J = 8.7 Hz, Ph), 7.85 (d, 4H, J = 8.6 Hz, Ph), 7.45 (m, 2H, Ph), 7.41 (m, 2H, Ph), 7.01 (d, 4H, J = 8.89 Hz, Ph), 5.87–5.82 (m, 2H), 5.04 (dd, 2H, J = 13.6 Hz), 4.95 (dd, 2H, J = 9.6 Hz), 4.04 (t, 4H, J = 6.5 Hz, OCH2–), 2.10–2.06 (m, 4H, –CH2–), 1.86–1.81 (m, 4H, –CH2–), 1.73–1.68 (m, 4H, –CH2–). δC(125 MHz; CDCl3; Me4Si) 26.92, 28.91, 30.23, 67.69, 109.25, 110.47, 121.57, 122.45, 124.51, 125.46, 126.13, 131.45, 136.54, 146.65, 152.36, 156.23, 162.34, 164.33.
2.5 1,2-Phenylene bis[4-{[4-(hept-6-en-1-yloxy)phenyl]diazenyl} benzoate] (4d)
Compound 4d was synthesis from 3d according to the method described for 4a. Yield of 4d: 0.0475 g, 30%. IR, νmax/cm−1 3066 (CH2), 2928 (CH2), 2852 (CH2), 1732 (CO, ester), 1644 (CC, vinyl), 1602, 1489 (CC, aromatic), 1252, 1139, 1076 (C–O), 836 (C–H). δH(500 MHz; CDCl3; Me4Si) 8.23 (d, 4H, J = 8.7 Hz, Ph), 7.92 (d, 4H, J = 8.7 Hz, Ph), 7.86 (d, 4H, J = 8.5 Hz, Ph), 7.46 (m, 2H, Ph), 7.40 (m, 2H, Ph), 7.02 (d, 4H, J = 8.7 Hz, Ph), 5.85–5.80 (m, 2H), 5.04 (dd, 2H, J = 13.8 Hz), 4.92 (dd, 2H, J = 9.6 Hz), 4.04 (t, 4H, J = 6.5 Hz, OCH2–), 2.11–2.05 (m, 4H, –CH2–), 1.86–1.80 (m, 4H, –CH2–), 1.82–1.77 (m, 4H, –CH2–), 1.74–1.67 (m, 4H, –CH2–). δC(125 MHz; CDCl3; Me4Si) 25.04, 26.35, 27.45, 30.32, 67.67, 109.66, 110.89, 121.35, 122.38, 124.29, 125.55, 126.23, 131.55, 136.47, 146.63, 152.34, 156.21, 162.14, 164.45.
2.6 1,2-Phenylene bis[4-{[4-(oct-7-en-1-yloxy)phenyl]diazenyl} benzoate] (4e)
Comp. 4e was synthesis from 3e according to the method described for 4a. Yield of 4e: 0.1251 g, 35%. IR, νmax/cm−1 3076 (CH2), 2925 (CH2), 2855 (CH2), 1735 (CO, ester), 1640 (CC, vinyl), 1602, 1500 (CC, aromatic), 1261, 1141, 1060 (C–O), 841 (C–H). δH(500 MHz; CDCl3; Me4Si) δ: 8.24 (d, 4H, J = 8.7 Hz, Ph), 7.92 (d, 4H, J = 8.9 Hz, Ph), 7.84 (d, 4H, J = 8.6 Hz, Ph), 7.45 (m, 2H, Ph), 7.41 (m, 1H, Ph), 7.01 (d, 4H, J = 8.6 Hz, Ph), 5.86–5.80 (m, 2H), 5.05 (dd, 2H, J = 13.7 Hz), 4.94 (dd, 2H, J = 9.6 Hz), 4.04 (t, 4H, J = 6.5 Hz, OCH2–), 2.08–2.02 (m, 4H, –CH2–), 1.86–1.81 (m, 4H, –CH2–), 1.74–1.66 (m, 8H, –CH2–). δC(125 MHz; CDCl3; Me4Si) 24.94, 26.24, 30.78, 32.34, 33.40, 68.19, 114.84, 114.89, 122.63, 122.70, 125.12, 125.37, 127.62, 131.46, 138.40, 146.69, 151.90, 156.71, 162.50, 164.67.
2.7 Instruments
The structure of the compounds was confirmed by spectroscopic method. IR spectra were recorded with a Perkin Elmer FTIR spectrometer (670). 1H NMR (500 MHz) and 13C NMR (125 MHz) spectra were recorded with a Bruker (DMX500) spectrometer. The transition temperatures and their enthalpies were measured by differential scanning calorimetry (Perkin DSC 7) with heating and cooling rates were 10 °C min−1. Optical textures were obtained by using Olympus BX51 polarizing optical microscope attached with Olympus DP26 digital camera equipped with a Mettler Toledo FP82HT hot stage and a FP90 central processor unit. X-ray diffraction measurements were carried out using Cu-Kα radiation (λ = 1.54 Å) generated from a 4 kW rotating anode generator (Rigaku Ultrax-18) equipped with a graphite crystal monochromator. Sample was filled in Hampton research capillaries (0.5 mm diameter) from isotropic phase, sealed and held on a heater. X-ray diffraction was carried out in the mesophase obtained on cooling the isotropic phase. Though a magnetic field of about 5k Gauss was used to align the samples, the diffraction patterns indicate that the sample was not aligned perfectly and, therefore, should be considered as unaligned sample. Absorption spectra for photochromic study were recorded using an Ocean Optics HR2000 + miniature UV-Vis spectrophotometer. All the solutions were prepared and measured under air in the dark at room temperature (21 ± 1 °C) using 1 cm quartz cells. The cells were closed to avoid the evaporation of the solvent and the solutions were stirred during the irradiation time. The solutions were irradiated at λmax = 365 nm along with heat filter to avoid any extra heat radiation to the sample. The UV light illumination is performed using Omnicure S2000 UV source with variable intensities (5–10 mW cm−2).
3. Results and discussion
3.1 Synthesis
The synthesis of compounds 4a–e was performed as depicted in Scheme 1. The azobenzene containing rod-like side arms were synthesized by a literature procedure.22,24 The diazonuim salt was prepared with sodium nitrite and subsequent coupling with phenol to afforded the ethyl 4-[(4-hydroxyphenyl)diazenyl]benzoate 1 which is purified by crystallization and recrystallization from methanol in about 51% yield. Compound 1 was alkylated with 4-bromo-1-butene to give the ethyl 4-{[4-(but-3-en-1-yloxy)phenyl]diazenyl}benzoate 2a, which was purified by column chromatography on silica followed by crystallization from methanol/chloroform in about 60% yield. Other compounds (2b–e) were also synthesized from the corresponding bromoalkene with the same method of compound 2a in good overall yield up to 77% for compound 2c. Compounds 2a–e were hydrolysed under basic conditions to yield the 4-{[4-(alkyl-en-1-yloxy)phenyl]diazenyl} benzoic acid 3a–e. Hence, compound 4-{(E)-2-[4-(but-3-en-1-yloxy)phenyl]diazen-1-yl}benzoic acid 3a was given a nice crystals from ethanol/chloroform mixture. For a series of U-shaped compounds, acids 3a–e were coupled with 1,2-dihydroxybenzene by DCC and DMAP to achieved the corresponding desired molecules 4a–e (Scheme 1), all compounds were purified by column chromatography on silica gel and recrystallization. All compounds were characterized using 1H, 13C NMR and elemental analysis. Analytical data were found to be in good agreement with the structures (see detailed synthetic procedures and analytical data in ESI†).
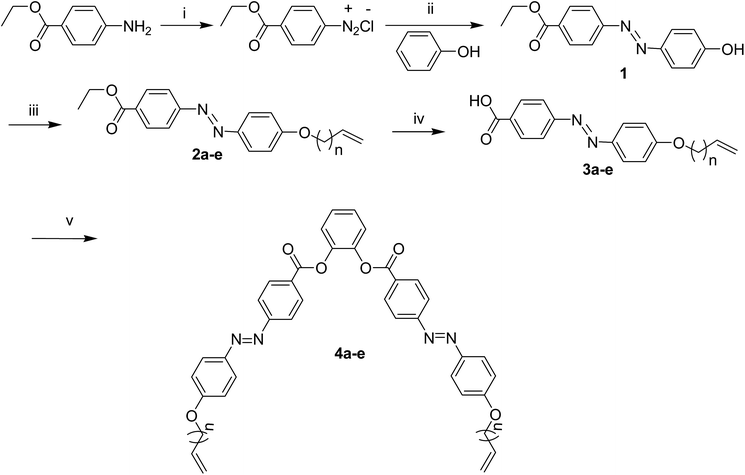 |
| | Scheme 1 Synthesis of compounds 4a–e. Reagents and conditions: (i) NaNO2, 3 equivalent HCl, 2 °C; (ii) phenol, NaOH, pH 9, 2 °C; (iii) K2CO3, KI, BrCnH2n−1 (n = 2–6), reflux; (iv) KOH, MeOH, reflux; (v) catechol, DCC, DMAP. | |
3.2 Mesomorphic properties
The liquid crystalline properties were studied by polarized-light optical microscopy (POM), differential scanning calorimetry (DSC) and X-ray diffraction analysis. DSC studies confirmed the phase transition temperatures (T/°C) observed by polarizing microscopy and gave the enthalpy changes [ΔH/J g−1] associated with these phase transitions as shown in Table 1.
Table 1 Phase transition temperature (T/°C) and associated transition enthalpy values [ΔH/J g−1] observed for the second heating and cooling DSC scans of 4a–ea
| Compound |
n |
Heating |
Cooling |
| Peak temperatures from DSC (rate 10 °C min−1); abbreviation Cr = crystal, SmA = smectic A phase, I = isotropic phase. |
| 4a |
2 |
Cr 101 [30] SmA 160 [14] I |
I 155 [14] SmA 90 [24] Cr |
| 4b |
3 |
Cr 98 [30] SmA 156 [14] I |
I 151 [14] SmA 85 [27] Cr |
| 4c |
4 |
Cr 93 [23] SmA 154 [12] I |
I 150 [12] SmA 80 [22] Cr |
| 4d |
5 |
Cr 111 [30] SmA 152 [12] I |
I 148 [12] SmA 97 [25] Cr |
| 4e |
6 |
Cr 81 [17] SmA 146 [8] I |
I 144 [8] SmA 74 [16] Cr |
3.2.1 DSC-studies. For the series of U-shaped compounds 4a–e, having even and odd number of alkyl carbons the enantiotropic phase sequence Cr–SmA–I is observed for all compounds, independent on the chain parity. It was observed that the melting points are reduced from compound 4a to 4e, those exhibits the enantiotropic SmA phases. On cooling cycle, the transition temperatures are lower than those of the lower homologues compound, for example 4a. Also the SmA–I transition enthalpy values decrease with growing chain length as presented in Table 1. The DSC curves for all compounds 4a–e are shown in Fig. 1 and DSC graphs with full details are presented in the ESI (Fig. S1†).
 |
| | Fig. 1 DSC scans of second heating ( ) and cooling ( ) and cooling ( ) curves of compound 4a–e (n = 2–6) at 10 °C min−1. ) curves of compound 4a–e (n = 2–6) at 10 °C min−1. | |
3.2.2 Polarizing optical microscopy (POM) studies. All the U-shaped compounds (4a–e) derived from 1,2-dihydroxybenzene show fan shaped textures, typical for smectic phases, as presented in Fig. 2. In all cases the extinction crosses are parallel to polarizer and analyzer which indicate nontilted smectic phases. Upon shearing homeotropic alignment is immediately achieved; these homeotropically aligned regions are completely isotropic down to the crystallization temperature, confirming the presence of a uniaxial SmA phases for these compounds. The typical textures of 4a and 4b are shown in Fig. 2, additional textures for 4b, 4c and 4d are presented in the ESI (Fig. S2†). All the transition temperatures observed under POM were matching with DSC data. The phase structures were further characterized by X-ray diffraction analysis for a selected example of each phase type.
 |
| | Fig. 2 Polarized optical microphotographs of texture: (a) SmA phase at 124 °C of 4a and (b) SmA phase at 120 °C of 4b. | |
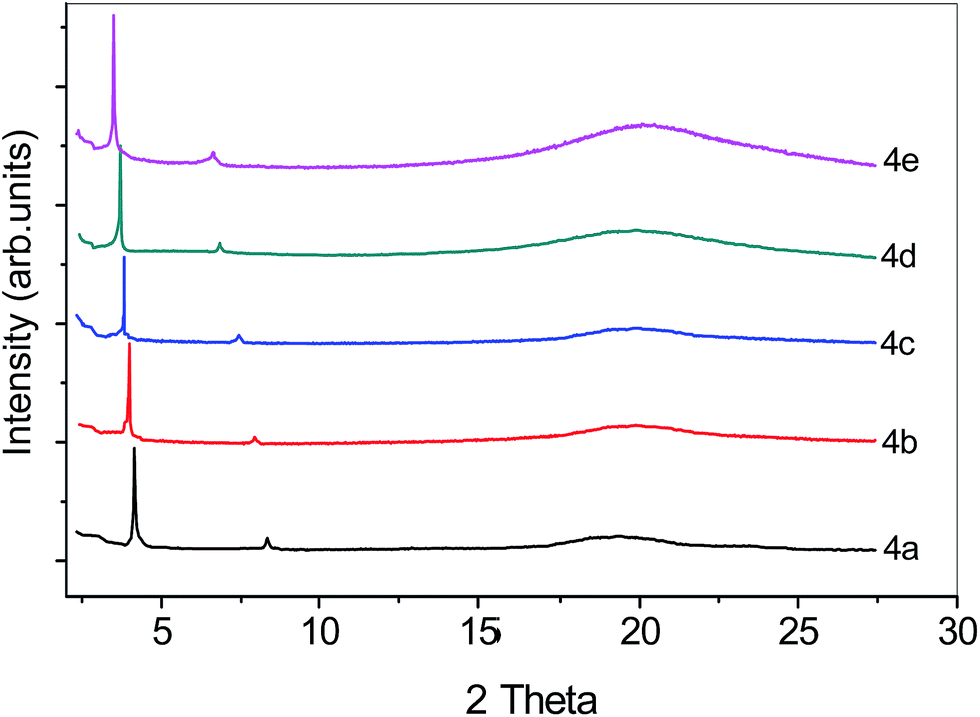
3.2.3 X-ray diffraction studies. The X-ray diffraction studies confirm the phase assignment. X-ray diffraction was carried out in the mesophases obtained on cooling from the isotropic phases. The intensity versus 2θ plot derived from the diffraction pattern of the compounds 4a–e (n = 2–6) are shown in Fig. 3. The diffraction pattern of all compound 4a–e exhibits a sharp reflection in the small angle region, corresponding to d = 21.12–26.60 Å, which is nearly equal to the corresponding molecular length of 4a–e (ranging from 21.10–26.49 Å) as estimated from top to bottom of a U-shaped conformer shown in Fig. 4a and estimated lengths are presented in Table 2. The weak small reflection at d = 10.41–13.27 Å is the second order of the layer reflex. In the wide-angle region a diffuse peak at d ≈ 4.22–4.66 Å confirms a fluid smectic phase without in-plane order. Therefore, we anticipate that compound 4a–e exhibits a smectic A type phase. This is most likely due to the V-shape of the molecules 4 which favours a parallel organization of the adjacent rod-like azobenzene units (thus adopting a U-shaped conformation).
 |
| | Fig. 3 Intensity versus 2θ graph derived from the X-ray diffraction for the SmA phase of compound 4a–e at 124, 120, 118, 114 and 110 °C, respectively. | |
 |
| | Fig. 4 (a) Major conformation of compound 4a–e and (b) organization of these molecules in the SmA phase. | |
Table 2 X-ray Diffraction data for compound 4a–e
| Compound |
l, Å |
θ° |
d-spacing, Å (hk) |
| 4a |
21.10 |
2.09 |
21.12 (01) |
| 4.25 |
10.41 (02) |
| 9.50 |
4.66 (halo) |
| 4b |
22.36 |
1.96 |
22.42 (01) |
| 3.97 |
11.14 (02) |
| 9.58 |
4.61 (halo) |
| 4c |
23.72 |
1.85 |
23.78 (01) |
| 3.77 |
11.72 (02) |
| 9.85 |
4.57 (halo) |
| 4d |
25.06 |
1.75 |
25.10 (01) |
| 3.41 |
12.51 (02) |
| 9.75 |
4.54 (halo) |
| 4e |
26.49 |
1.65 |
26.60 (01) |
| 3.33 |
13.27 (02) |
| 10.51 |
4.22 (halo) |
However, the alignment of the rod-like cores cannot be fully parallel, so that some wedge-shape (U-shaped conformation) of the molecules is retained which requires an antiparallel packing of the molecules with intercalated (mixed) aliphatic chains and azobenzene cores to optimize the space filling; the catechol groups from their own layers and separate the layers formed by the rod-like units and terminal chains, as shown in Fig. 4b. A different packing pattern is proposed by X-ray diffraction analysis from the smectic A phase formed by U-shaped compounds. A layer spacing of d = 53.4 Å was observed whereas the length of the molecule (l) including alkyl chains in the all trans conformation was estimated to be ≈31 Å. The ratio d/l ≈ 1.72 suggests that in this smectic A phase the molecules are ordered into bilayers.16 However, similar packing patterns were observed for some compounds.15 The X-ray diffraction suggested that the layer spacing is about the same as the length of the molecule in a U-shaped. The XRD result suggests that the smectic A phase is a monolayer structure in which molecules can exist in a U-shaped.15 Thus, depending on bent unit (1,2-dihydroxybenzene) and the parity of chain length three distinct types of LC phases were observed for these non-linear azobenzene based mesogens. The distinct phase types seem to result from distinct molecular conformations to which the molecules adopt in the self-assembly process in order to optimize their packing.
3.3 Photo-switching study
The photo-switching studies were initially carried out on solutions and then on liquid crystal cells. Consequently the materials behavior with respect to UV light is obtained and also these results are indispensable for creating optical storage devices.
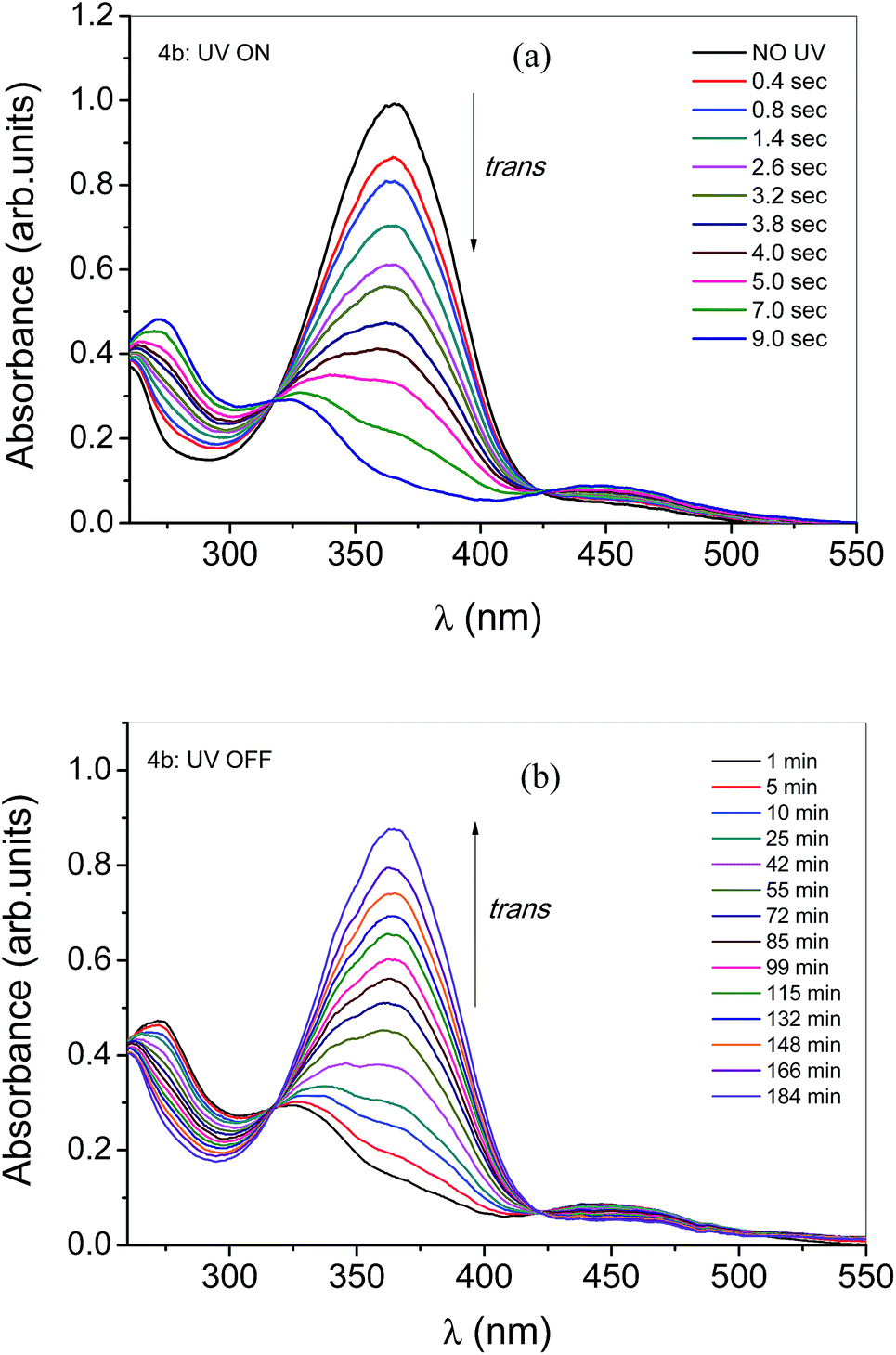
All U-shaped molecules also showed similar absorption spectra due to their similar molecular structure with variable alkyl chain (n = 2–6). Fig. 5 depicts the absorption spectra of 4a (n = 2) before and after UV illumination (see spectra of 4b–e in Fig. S3 in the ESI†). The absorption spectra of compound 4a show absorbance maxima at 364 nm. The absorption spectra of compound was carried out also in chloroform solution having same concentration C = 1.2 × 10−5 mol L−1. Compound 4a was illuminated with UV light with 365 nm filter at different time intervals and promptly absorption spectra were recorded. The absorption maximum at 364 nm decreases due to E/Z photoisomerization due to E isomer transformed to Z isomer. After ∼14 s illumination, there is no much change in absorption spectrum confirms the saturation of E/Z isomerization process.
 |
| | Fig. 5 Spectra shows the absorbance behavior of the compound 4a when UV light is shined on the sample. It is evident that within ∼15 seconds of illumination, photo saturation occurs. Data were taken before shining UV light (no UV) and with subsequent time intervals (light intensity at 5 mW cm−2). | |
Fig. 6 shows the E–Z absorption of compound 4a–e as a function of exposure time. Data is extracted from Fig. 5 (and Fig. S3 in the ESI†). The wavelength is fixed at 364 nm and absorption values were recorded as a function of exposure time with UV intensity being fixed at 5 mW cm−2. Curve shows that photosaturation occurs within 15 s for 4a and photosaturation time for other compounds 4b–e shows ranging from 14–15 s, which is faster as compared with nematic to isotropic phase involved photoisomerization.36
 |
| | Fig. 6 Photoisomerization curve of 4a–e as a function of UV illumination time showing trans to cis behaviour (light intensity at 5 mW cm−2). | |
Fig. 7 shows the thermal back relaxation process where the solution is shined continuously for 20 seconds (photo stationery state) and kept in the dark and then at subsequent time intervals, spectral data were recorded. Thermal back relaxation for the compound 4a showed in Fig. 7 (Fig. S4 for 4b–e in the ESI†).
 |
| | Fig. 7 Thermal back relaxation process for the compound 4a shows that to relax from cis to trans takes around 235 min. Solution is shined for 20 seconds (photo stationery state) and then at subsequent intervals data were recorded (light intensity at 5 mW cm−2). | |
Fig. 8 shows the time dependence of the Z–E absorption of compound 4a–e. Peak wavelength at 364 nm as obtained from Fig. 7 (and Fig. S4 in the ESI†) is plotted as a function of recovery time. The thermal back relaxation occurs 235, 362, 322, 380, 350 minutes were time taken to relax back to their original state for the compounds 4a–e, respectively, within this time, it is reasonably fast as compared with nematic to isotropic phase involved thermal back relaxation.36
 |
| | Fig. 8 Photoisomerization curve of Z isomer (4a–e) as a function of recovery time when UV light is illuminated until it reaches photosaturation and followed by measuring back relaxation time (light intensity at 5 mW cm−2). | |
Prasad et al.36 reported that the faster thermal back relaxation is due to their layered structure since changes are confined to in-plane rotation of the molecules as compared with nematic to isotropic phase involve transition. This hypothesis is well-established by the fact that a similar feature was observed in another case wherein the two phases involved have a layer structure.37
3.3.1 Kinetic study. We also calculated the rate constant (kt) for the cis–trans isomerization behaviour at room temperature for all five compounds according to the eqn (1) reported by Lutfor et al.25| |
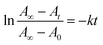 | (1) |
Where At, A0 and A∞, is the absorbance at 365 nm of time t, time zero and infinite time, respectively. A typical first order plot using eqn (1) at room temperature (25 °C) for all five compounds are shown Fig. 9. A typical first order behaviour for 4a–e shows throughout the relaxation time. All compounds shows similar behaviour at room temperature irrespective of their terminal alkyl chains length. Apparently, all compounds showed first order exponential decay in solutions. The rate constants were observed for the Z–E isomerization of 7.92 × 10−3, 5.60 × 10−3, 6.91 × 10−3, 7.30 × 10−3 and 5.28 × 10−3 s−1 for 4a–e, respectively. The rate constants are not substantial variation with respect to the alkyl chain length (n = 2–6).
 |
| | Fig. 9 First order plots for the compounds 4a–e for cis–trans isomerization. Conditions: c = 1.2 × 10−5 mol L−1 in chloroform at room temperature (25 °C) (light intensity at 5 mW cm−2). | |
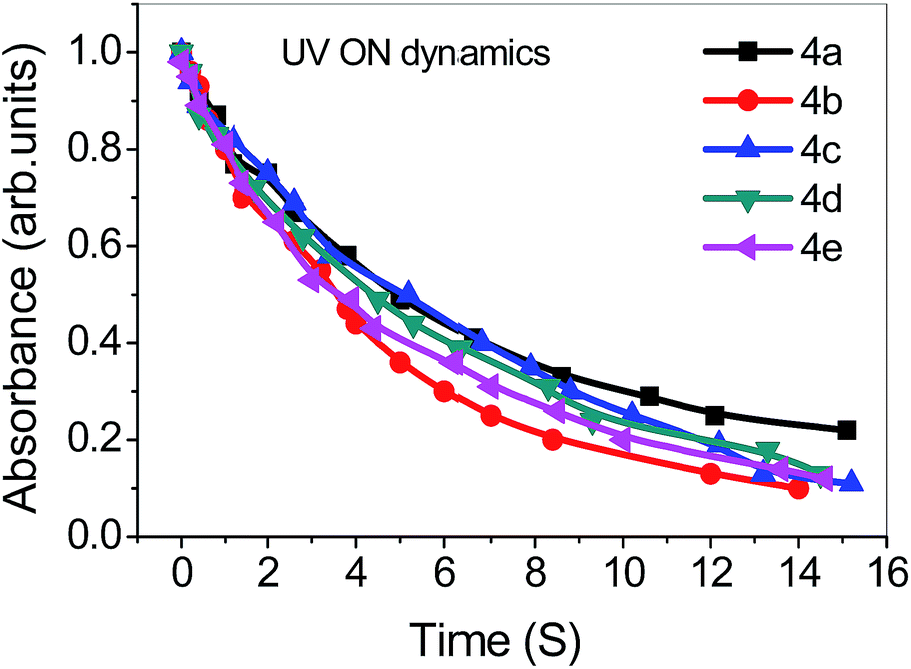
3.3.2 Effect of alkyl chain length. For U-shaped molecular architectures, we used alkyl chains as n = 2–6, we found little odd-even effect on the photoisomerization of UV light with intensity at 5 mW cm−2 for compounds 4a–e which is shown in Fig. S5.† The UV on dynamics to the U-shaped molecules showed small variation such as 4a = 15.1, 4b = 14.0, 4c = 15.2, 4d = 14.5 and 4e = 14.6 s. Thus, a little odd–even effect is seen on the U-shaped compounds. The odd–even effect on the photoisomerization of back light for compounds 4a–e is also shown in Fig. S6.† The UV off dynamics to the U-shaped showed substantial variation for 4a = 235, 4b = 362, 4c = 333, 4d = 380 and 4e = 350 minutes. For U-shaped compounds all exhibit high thermal back relaxation. Presented study is a strong evidence for odd–even effect when no light is shined on them.
3.3.3 Effect of light intensity. The photoisomerization of compound 4b was studied with variable intensities (5, 10 and 15 mW cm−2). Fig. 10 shows the photoisomerization behaviour of 4b using UV light with intensity at 10 mW cm−2. The photosaturation occurs faster 9 s when intensity of UV light is increased (Fig. 10a) and the thermal back relaxation was also fast 184 min as shown in Fig. 10b. In addition, we have studied the photoisomerization behaviour of 4b with more intensity at 15 mW cm−2. The photosaturation occurs very fast within 7 s only and the thermal back relaxation was also very fast 144 min as shown in Fig. S7 at ESI.† Thus, the time intervals of photosaturation and thermal back relaxation are decreased when intensity of the irradiated light is increased. Therefore, the intensity is inversely proportional to the time of photosaturation and thermal back relaxation (Table 3).
 |
| | Fig. 10 (a) Absorption spectra of 4b UV light with intensity at 10 mW cm−2 and (b) thermal back relaxation process for 4b. | |
Table 3 Photoisomerization of 4b with variable intensities
| Intensity (mW cm−2) |
Photosaturation (s) |
Back relaxation (min) |
| 5.0 |
14 |
362 |
| 10.0 |
9 |
184 |
| 15.0 |
7 |
144 |
3.3.4 Photo-stability test. Multiple cycles of photoisomerization is considered here, irrespective of time of trans–cis and cis–trans isomerization. Fig. 11 shows the photo-stability of the light sensitive compound 4a and 4b. Both compounds are used the chloroform solvent with the concentration 1.1 × 10−5 mol L−1. The UV light of intensity 10 mW cm−2 was illuminated on the solution continuously, until photo-stationary state is reach. Immediately, visible light of 10 mW cm−2 (450 nm wavelength) was irradiated on the solution for back relaxation process after photosaturation state. This phenomenon was continued 4 times to evaluate the photo-stability of the compound. Thus, compound is stable towards light and does not degrade via light illumination. The extent of reversible isomerization did not significantly decay after 4 cycles, indicating that the photo-responsive properties of both compounds were stable and repeatable. Therefore, these U-shaped liquid crystals are suitable in optical storage device industries, due to the excellent light induced characteristics.
 |
| | Fig. 11 Reversibility of the photoisomerization process of the azobenzene chromophore of 4a and 4b in chloroform with the concentration of 1.1 × 10−5 mol L−1. | |
Spectral investigation on solid films of 4a was used as a representative compound and data were also recorded as a function of UV illumination. Here guest–host effect is employed where 5CB, room temperature nematic liquid crystal is act as a host and bent-shaped liquid crystals are act as guest systems. Previously prepared polyimide coated, unidirectionally rubbed sandwiched cells were filled with the guest–host mixture at isotropic temperature of the mixture (∼50 °C). UV/Vis spectral data were recorded using spectrophotometer.
Fig. 12a shows the absorbance versus wavelength graph, subjected to before and after shining UV radiation of wavelength 365 nm. It is observed that, within 4 seconds of illumination, system attains photosaturation state. Whereas in Fig. 12b, thermal back relaxation for the solid cells were captured after illuminating the cells for photosaturation state (around 4 seconds) and then at subsequent intervals spectral data were recorded. To be comparison, no UV point is also given. So it is clear from the graph that, system takes around 70 minutes of time to relax back to the original configuration.
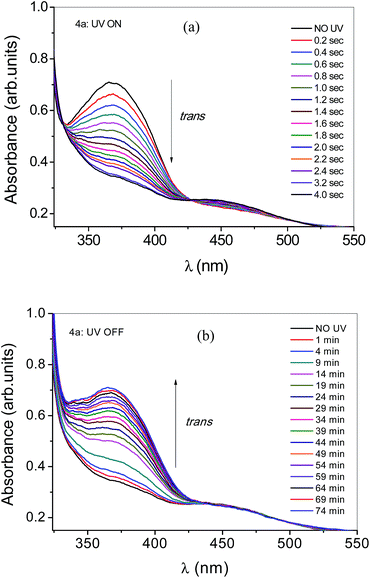 |
| | Fig. 12 (a) Absorption spectra of 4a in solid with different exposure time of UV light. No UV corresponds to the 0 seconds UV light illumination. (b) Thermal back relaxation process for the compound 4a after illuminating the material for 5 seconds (photo stationery state). | |
The dynamics of UV on and UV off (thermal back relaxation) process for the compound 4a is shown in Fig. 13. Peak wavelength is 364 nm and data is generated from peak absorbance at 364 nm as a function of exposure time (Fig. 13a). It was clearly observed that E–Z conversion takes around 4 seconds whereas recovery to the original state i.e., thermal back relaxation takes around 74 minutes. In Fig. 13b, No UV point is given to make it clear that system already reached its original value. As compare to liquid, solid behaviour is fast may be due to the tightly packed molecules in cells whereas in solutions, molecules are having more freedom to move around. The isothermal phase transition of LCs in the LC cell was created with the increase of the population of the cis-azo-LCs.38 Certainly the unstable cis-azo-LCs can be recovered back to the stable trans-azo-LCs by illumination with green light and the transformation rate from cis- to trans-isomers is much higher than that of dark relaxation.39 Kundu et al.40 demonstrated in situ homeotropic alignment by photochromic trans- to cis- isomerization of the azo-dye doped in a nematic host. The augmented dipole moment of a cis-isomer formed under UV-irradiation expedites a molecular assembly into crystalline aggregates. Subsequent deposition of the aggregates creates roughened surface and induces anchoring transition from the initial planar to homeotropic alignment of LCs. The alignment is unwavering against temperature, light and chemical treatment which practically permanent for a device implementation.
 |
| | Fig. 13 Photo-isomerization curve of E isomer (4a) showing UV on process (a) and UV off process (b) as a function of time. One can see that conversion from E/Z occurs at around 4 seconds whereas Z/E occurs around 74 minutes. | |
To demonstrate the potential of the materials stated here, an optical storage device (Fig. 14) that is realized by using the above mentioned method. The mixture is capillary filled into the commercially available cell (Instec) ITO + polyimide coated, unidirectional rubbed, sandwiched cell at isotropic temperature (∼70 °C). Qualities of the cells were observed under optical polarizing microscope. The guest–host mixture was illuminated with UV light of 10 mW cm−2 intensity through a standard mask for 10 minutes. The blue-green region is the area which is masked with UV radiation which remains in liquid crystalline state whereas the grey region in the central position is the erasing area which is illuminated with UV radiation which transforms to isotropic state.
 |
| | Fig. 14 Optical data storage device based on the principle described in this article observed under the crossed polarizers. The sample was kept at room temperature and illuminated with UV radiation through a photo masks. The colour erasing region in the middle position is the molecules which are exposed to UV radiation and the blue-green region where the radiation is masked. | |
As expected material transforms from order to disorder state with the illumination of light giving high contrast between blue-green and grey states (erased in the middle). Research is in progress to stabilize these materials to use it as permanent optical storage devices by incorporating polymeric chains to photopolymerize the structure.
4. Conclusion
Five new U-shaped mesogens having azobenzene units are reported in this paper. The double bonds can be used for preparation of polymers or silylfunctionalized U-shaped core mesogens, whereas the presence of the azo linkage in these liquid crystal monomers is suitable for photochromism studies. Thus, U-shaped all monomers showed stable enantiotropic monolayer SmA type phase. The photoswitching properties of compounds 4a–e of this series shows trans to cis isomerization around 14–15 s, and reverse process take place ranging from 235–380 min in solutions whereas in solids state E–Z photoisomerization of 4a takes around 4 s and reverse back to original Z–E takes around 74 min. Under UV light irradiation in solution, all compounds (4a–e) behave first order rate law throughout the relaxation time and the substantial effect for cis to trans is found on the odd-even chain length. The photosaturation and thermal back relaxation times are decreased when intensity of the irradiated light is increased. The photo-responsive properties are found stable and repeatable due to the compounds did not degrade on light illumination at 10 mW cm−2 with multiple cycles. Thus, the photoswitching behavior of these materials may be suitably exploited in the field of optical data storage device and in molecular switches which needs fast switching as well for permanent optical storage devices. Indeed, so far, hardly any azo compound is reported which exhibits such a fast switching property in solid state.
Acknowledgements
This research was supported by PRGS research grant (RDU 130803). Thank Mrs. K. N. Vasudha for supporting this work.
Notes and references
- V. C. Yelamaggad, I. Shashikala, S. D. Rao Shankar and S. P. Krishna, Liq. Cryst., 2004, 31, 1027–1036 CrossRef PubMed.
- S. Chandrasekhar, B. K. Sadashiva and K. A. Suresh, Pramana, 1997, 9, 471–480 CrossRef.
- S. Chandrasekhar, Liquid Crystals, Cambridge University Press, Cambridge, 2nd edn, 1994 Search PubMed.
- G. P. Gennes De and J. Prost, The Physics of Liquid Crystals, Oxford Science Publication, Oxford, 1993 Search PubMed.
- C. Tschierske, J. Mater. Chem., 1998, 8, 1485–1508 RSC.
- C. Tschierske, J. Mater. Chem., 2001, 11, 2647–2671 RSC.
- C. Tschierske, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2001, 97, 191–267 RSC.
- C. Tschierske, Curr. Opin. Colloid Interface Sci., 2002, 7, 69–80 CrossRef CAS.
- D. Vorlander and A. Apel, Ber. Dtsch. Chem. Ges., 1929, 62, 2831 CrossRef PubMed.
- D. Vorlander and A. Apel, Ber. Dtsch. Chem. Ges., 1932, 65, 1101 CrossRef PubMed.
- G. Pelzl, I. Wirth and W. Weissflog, Liq. Cryst., 2001, 28, 969–972 CrossRef CAS PubMed.
- M. Kuboshita, Y. Matsunaga and H. Matsuzaki, Mol. Cryst. Liq. Cryst., 1991, 199, 319–326 CrossRef CAS PubMed.
- D. Demus, Liq. Cryst., 1989, 5, 75–110 CrossRef CAS PubMed.
- G. Pelzl, S. Diele and W. Weissflog, Adv. Mater., 1999, 11, 707–724 CrossRef CAS.
- A. Yamaguchi, M. Watanabe and A. Yoshizawa, Liq. Cryst., 2007, 34, 633–639 CrossRef CAS PubMed.
- S. G. Attard and G. A. Douglass, Liq. Cryst., 1997, 22, 349–358 CrossRef PubMed.
- M. R. Lutfor, T. K. Biswas, S. M. Sarkar, M. M. Yusoff, M. Fazli Abdul Malek and C. Tschierske, J. Mol. Liq., 2015, 202, 125–133 CrossRef PubMed.
- H. Matsuzaki and Y. Matsunaga, Mol. Cryst. Liq. Cryst., 1993, 14, 105–120 CAS.
- T. Kato, H. Adachi, A. Fujishima and J. M. J. Frechet, Chem. Lett., 1992, 265–268 CrossRef CAS.
- M. Tanaka and A. Yoshizawa, J. Mater. Chem. C, 2013, 1, 315–320 RSC.
- N. V. S. Rao and M. Kr. Paul, Mol. Cryst. Liq. Cryst., 2003, 393, 57–66 CrossRef CAS PubMed.
- M. R. Lutfor, H. Gurumurthy, Y. M. Mashitah, T. H. Srinivasa, S. A. Nurlin, M. A. M. Nor Fazli and S. Kumar, New J. Chem., 2013, 37, 2460–2467 RSC.
- M. R. Lutfor, H. Gurumurthy, A. Mahrokh, Y. M. Mashitah and K. Sandeep, J. Fluorine Chem., 2013, 156, 230–235 CrossRef PubMed.
- M. R. Lutfor, A. Jahimin, K. Sandeep, S. Silong and M. Z. A. Rahman, Phase Transitions, 2009, 82, 228–239 CrossRef PubMed.
- M. R. Lutfor, M. M. Yusoff and S. Kumar, RSC Adv., 2014, 4, 35089–35098 RSC.
- T. Sasaki, T. Ikeda and K. Ichimura, J. Am. Chem. Soc., 1994, 116, 625–628 CrossRef CAS.
- A. Stracke, J. H. Wendorff, D. Goldmann and D. Janietz, Liq. Cryst., 2000, 27, 1049–1057 CrossRef CAS PubMed.
- M. Eich and J. Wendorff, J. Opt. Soc. Am. B, 1990, 7, 1428–1436 CrossRef CAS.
- T. Urbas, V. Tondiglia, L. Natarajan, R. Sutherland, H. Yu, H. J. Li and T. Bunning, J. Am. Chem. Soc., 2004, 126, 13580–13581 CrossRef PubMed.
- K. Ichimura, K. S. Oh and M. Nakagawa, Surf. Sci., 2000, 288, 1624–1626 CAS.
- L. Komitov, C. Ruslim, Y. Matsuzawa and K. Ichimura, Liq. Cryst., 2000, 27, 1011–1016 CrossRef CAS PubMed.
- T. Ikeda and O. Tsutsumi, Science, 1995, 268, 1873–1875 CAS.
- T. Ikeda, S. Horiuchi, B. D. Karanjit, S. Kurihara and S. Tazuke, Macromolecules, 1990, 23, 36–42 CrossRef CAS.
- M. R. Lutfor, G. Hegde, S. Kumar, C. Tschierske and G. V. Chigrinov, Opt. Mater., 2009, 32, 176–183 CrossRef CAS PubMed.
- T. Ikeda, J. Mater. Chem., 2003, 13, 2037–2057 RSC.
- S. K. Prasad, K. L. Sandhya, G. G. Nair and D. S. Shankar Rao, Curr. Sci., 2004, 86, 815–823 CAS.
- S. K. Prasad, K. L. Sandhya, D. S. Shankar Rao and Y. S. Negi, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 67, 051701/1 CrossRef CAS.
- A. Y. G. Fuh, Y. C. Liu, K. T. Cheng, C. K. Liu and Y. D. Chen, Sci. Adv. Mater., 2014, 6, 37–42 CrossRef CAS PubMed.
- Y. C. Liu, K. T. Cheng, Y. D. Chen and A. Y. G. Fuh, Opt. Express, 2013, 21, 18492–18500 CrossRef PubMed.
- S. Kundu, M. H. Lee, S. H. Lee and S. W. Kang, Adv. Mater., 2013, 25, 3365–3370 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, representative DSC graphs, POM and UV/vis absorption spectra. See DOI: 10.1039/c5ra12705j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 


 ) and cooling (
) and cooling ( ) curves of compound 4a–e (n = 2–6) at 10 °C min−1.
) curves of compound 4a–e (n = 2–6) at 10 °C min−1.