
Palladium catalyzed C(sp2)–C(sp2) bond formation. A highly regio- and chemoselective oxidative Heck C-3 alkenylation of pyrones and pyridones†‡
Hafiz Ul Laha,
Faheem Rasoola and
Syed Khalid Yousuf*ab
aMedicinal Chemistry Division, Indian Institute of Integrative Medicine (CSIR-India), Srinagar-190005, India
bAcSIR-India, India. E-mail: khalidiiim@gmail.com; Fax: +91-194244133; Tel: +91-1942431253/+91-1942431255
First published on 10th September 2015
Abstract
Palladium catalysed ligand free direct dehydrogenative C-3 alkenylation of pyrones and unprotected pyridones from unactivated alkenes is reported. The process is highly regio- and chemoselective. A wide variety of alkene partners reacted to broaden the substrate scope of the process. The effect of the C-3 substituent on the rate of reaction is also studied.
The versatility of 2-pyrones and pyridones is evident by their record as structural and functional motifs of an innumerable number of natural products and bioactive molecules with diverse utilities.1 This has always inspired chemists towards the development of synthetically efficient strategies for their synthesis and structural diversifications to serve the purpose of harnessing their potential as useful candidates for medicinal chemistry.1,2 For structural diversifications C–C bond forming reactions including classical Heck/Suzuki coupling and Sonogashira reactions are well exploited with these motifs. All these reactions require predesigning of substrate like selective halogenation followed by subsequent functionalization making the library generation an intricate process. In recent years direct functionalization of C(sp2)–H bonds to generate new C–C bonds has emerged as a powerful tool in synthetic chemistry.3 Among the various C–C bonds forming reactions site selective C(sp2)–C(sp2) alkenylation has gained extraordinary importance both in synthetic organic and medicinal chemistry.4 This can be attributed to the fact that the so installed vinyl functionality can be exploited for accessing chemical diversifications using chemistry of choice. In case of pyrones and pyridones, there are only a few reports of oxidative C-alkenylation as witnessed by the literature.5 For example, Itahara applied direct C–H palladation and Heck alkenylation to N-methyl-2-pyridone, using acrylates to generate the C(5) adducts exclusively.6 Similarly, Georg et al. reported the palladium catalysed oxidative Heck alkenylation of dihydro-4-pyridone using methyl acrylates. Georg and co-workers described dehydrogenative alkenylation of cyclic enaminones via Fujiwara Moritoni reaction.7 However lack of studies towards regio- and chemoselectivity and pre-functionalisation of ring nitrogen are some of the concerns associated with these protocols. To the best of our knowledge there are no reports of oxidative C-alkenylation in case of 4-hydroxy 2-pyridones and other 4-substituted pyrones.
Utility of naturally occurring 6-styryl pyrones against infectious diseases such as trypanosomiasis, leishmaniasis, malaria or tuberculosis and their neuroprotective and hepatotoxicity is already well documented.8 However, the lack of diversity has always limited their use. This is because in pyrones and pyridones there are various sites like C-3, C-5 (in case of 4-hydroxy-6-methyl-2-pyrone) and N-1, C-3, C-5 (in case of 4-hydroxy-6-methyl-2-pyridone) liable for palladium activation (Fig. 1).
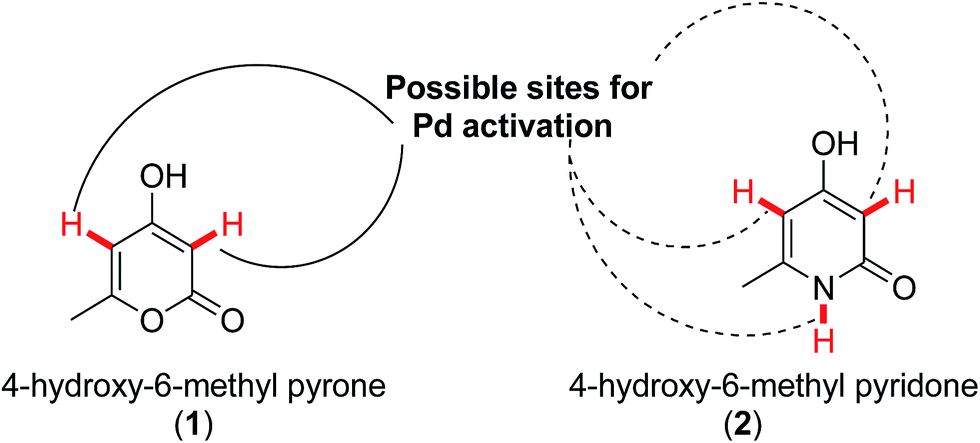 | ||
| Fig. 1 Possible sites for palladium activation in 4-hydroxypyrone (1) and pyridone (2). | ||
First-hand tactics for exploiting site-selectivity or facilitating the development of reagents that can supersede substrate predisposition to accomplish site-selective functionalizations seems to address these important restrictions and thereby have a foremost impact on small molecule science. With our continuous interest in developing new C–C bond forming reactions9 and in our research programme directed towards the synthesis of focused chemical libraries based on pyrone and pyridone skeleton as antituberculosis agents, we were driven for developing an efficient regioselective chemical strategy for the construction of various pyrone and pyridone derivatives bearing a functionalised vinyl unit at C-3 position keeping in view the medicinal importance of 6-steryl pyrones.
During our effort, we developed an efficient palladium catalysed protocol for the simple and facile oxidative C(sp2)–C(sp2) cross-coupling of various pyrones and pyridones with a range of activated/unactivated alkenes and herein we report the details of this study.
We started our preliminary study with 4-hydroxy-6-methyl-2-pyrone (1) and styrene for the dehydrogenative coupling to generate alkenylated product 3. A wide range of reaction parameters were examined to achieve the targeted product (Table 1). In all cases Pd(II) was used a catalyst with 5 mol% catalyst loading. Though use of Cu(OTf)2 and Pd(OAc)2 in different solvents under different conditions gave the expected product but the yield of the reaction was not satisfactory (Table 1, entry 1–8). Changing solvents from highly polar to moderate resulted in comparable or even lower yield. It was notable to find the dramatically high increase in the yield of the reaction using DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO (8:2) as solvent and Cu(OTf)2 as the oxidant in open air flask (Table 1, entry 9 and 10). In this case elevated temperature could only decrease the reaction time but there was no significant change in reaction yield. Prolonging the reaction time to 16 h in open air flask leads to the complete consumption of starting material at room temperature yielding the desired product in 75% yield (Table 1, entry 11). Use of other oxidants like Cu(OAc)2 AgCO3 could give the desired product albeit in lower yield. Abandoning the use of either oxidant or catalyst could not lead to the formation of any product. Thus, consolidating the outcome of our standardisation, we concluded that reaction of 4-hydroxy pyrone (1) (0.4 mmol, 1 equiv.) with styrene (0.44 mmol, 1.1 equiv.) in presence of Pd(OAc)2 (5 mol%), Cu(OTf)2 (5 mol%) in DMF/DMSO (8:2, 2 mL) solvent mixture in open air flask at room temperature for 16 h represents the most favourable set of conditions for the oxidative C-3 alkenylation.
:DMSO (8:2) as solvent and Cu(OTf)2 as the oxidant in open air flask (Table 1, entry 9 and 10). In this case elevated temperature could only decrease the reaction time but there was no significant change in reaction yield. Prolonging the reaction time to 16 h in open air flask leads to the complete consumption of starting material at room temperature yielding the desired product in 75% yield (Table 1, entry 11). Use of other oxidants like Cu(OAc)2 AgCO3 could give the desired product albeit in lower yield. Abandoning the use of either oxidant or catalyst could not lead to the formation of any product. Thus, consolidating the outcome of our standardisation, we concluded that reaction of 4-hydroxy pyrone (1) (0.4 mmol, 1 equiv.) with styrene (0.44 mmol, 1.1 equiv.) in presence of Pd(OAc)2 (5 mol%), Cu(OTf)2 (5 mol%) in DMF/DMSO (8:2, 2 mL) solvent mixture in open air flask at room temperature for 16 h represents the most favourable set of conditions for the oxidative C-3 alkenylation.
|
|||||
|---|---|---|---|---|---|
| Entry | Oxidantb | T (°C) | Solventc | Time (h) | Yieldd (%) |
| a In all cases 5 mol% of Pd(OAc)2 was used.b 5 mol% of Cu(OTf)2, Cu(OAc)2 and Ag2CO were used.c For 0.4 mmol of substrate, 2 mL of solvent was taken.d Isolated yield after column chromatography. | |||||
| 1 | Cu(OTf)2 | 100 | DMF | 8 | 55 |
| 2 | Cu(OTf)2 | rt. | DMF | 15 | 55 |
| 3 | Cu(OTf)2 | 100 | DMSO | 10 | 50 |
| 4 | Cu(OTf)2 | 100 | Dioxane | 10 | 35 |
| 5 | Cu(OTf)2 | 60 | DCM | 13 | 20 |
| 6 | Cu(OTf)2 | 80 | Toluene | 13 | 25 |
| 7 | Cu(OTf)2 | 80 | DCE | 10 | 20 |
| 8 | Cu(OTf)2 | 60 | CH3CN | 10 | 30 |
| 9 | Cu(OTf)2 | 80 | DMF:DMSO (8:2) |
8 | 70 |
| 10 | Cu(OTf)2 | rt. | DMF:DMSO (8:2) |
12 | 70 |
| 11 | Cu(OTf)2 | rt. | DMF:DMSO (8:2) |
16 | 75 |
| 12 | Cu(OTf)2 | rt. | DMF:DMSO (8:2) |
20 | 75 |
| 13 | Cu(OTf)2 | 80 | CH3NO2 | 8 | 35 |
| 14 | Cu(OAc)2 | 80 | DMF | 10 | 45 |
| 15 | Ag2CO3 | 80 | DMF | 7 | 10 |
| 16 | Ag2CO3 | rt. | DMF | 12 | 10 |
| 17 | None | 80 | DMF | 20 | 0 |

With these optimised reaction conditions in hand, a range of unactivated alkenes (Fig. 2) were allowed to react with 6-methyl-4-hydroxy-2-pyrone 1. Styrenes with both electron donating and electron with drawing groups reacted smoothly to generate corresponding C-3 alkenylated products in good to excellent yield. Position of substituent (o-, m- and p-) do not effect the reaction rate and yield. For example, ortho, meta and para methyl styrene reacted smoothly to yield their respective alkenylated products in good yield (Fig. 2, entries 8–10). It is worth mentioning that reaction of alpha methyl styrne afforded the corresponding oxidative Heck product 13 in 70% yield increasing the applicability of the process. Gratifyingly, 4-methoxy allyl benzene reacted smoothly to yield corresponding C-3 alkenylated product 14 in 70% yield. The fact that halo, nitro, alkyl and alkoxy groups were tolerated under the optimised reaction conditions expanded the substrate scope of the reaction. However acrylates failed to react with pyrone under the current reaction conditions.
 | ||
| Fig. 2 Substrate scope for dehydrogenative cross coupling of pyrone 1 and pyridone 2. | ||
Inspired by these positive results with pyrone 1, we extended our study to unprotected 4-hydroxy-6-methyl pyridone (2). Initially 2 was subjected to undergo the oxidative C3-alkenylation with styrene at room temperature (Table 1, entry 11). However, no conversion was found at room temperature even after 24 h of stirring. In this case heating the reaction mixture to 80 °C for 16 h lead to the complete conversion of 2 yielding the desired product 15 (Fig. 1, entry 15) in 70% yield with high regioselectivity. Although Itahara et al.,6 reported that N-methylpyridone and 4H-pyran-4-one are suitable substrates for dehydrogenative C–C bond formation with olefins. However in N-methylpyridone alkenylation occurs with competing C3- and C5-functionalization. Cheng and Gallagher10 extended oxidative cross-coupling to C6-substituted pyridones. To achieve regioselective alkenylation at C3, presence of an electron-donating substituent at C6 remained to be a deciding factor. It is noteworthy, that under the present conditions, no such prerequisite N-protection or structural designing was required. Encouraged by the results, next the oxidative alkenylation reaction in pyridone (2) was extended using a panel of styrenes to yield the corresponding C3-alkenylated pyridone derivatives (Fig. 2, entry 16–18) in good to high yield.
Based on the literature precedence on the directing effect of hydroxyl group in palladium catalysed oxidative C–C bond formation, we next tried to study the role of 4-OH in reaction reactivity and regioselectivity. In order to learn that, we subjected 4-methoxy-6-methyl-2-pyrone (19) to oxidative C-alkenylation reaction with styrene under optimised reaction conditions. It was observed that 19 reacted smoothly to yield the desired alkenylated pyrone 20 in 74% yield though only an elevated temperature of 80 °C in 10 h (Scheme 1).
 | ||
| Scheme 1 Cross dehydrogenative coupling of 4-methoxypyrone with styrene. | ||
Though the same regioselectivity was observed but the reactivity was decreased. Inspired by the above results and to further study the role of C-4 substituent, we became interested to investigate the fate of 4-chloro-2-pyrone (21) under the optimised reaction conditions. Accordingly, 21 was allowed to react with styrene at room temperature but there was no product formation even after 24 h. Thus, the reaction mixture was subjected to refluxing at 80 °C. The formation of product was witnessed after 16 h which was confirmed through spectroscopic analysis. As shown in Fig. 3, with several styrenes including both simple, electron donating and electron withdrawing, chloropyrone 21 afforded the corresponding C-3 alkenylated products with high level of regio- and chemoselectivity without formation of any classical Heck product. To the best of our knowledge it is the first report of chemoselective C(sp2)–C(sp2) oxidative alkenylation of chloropyrones. Such pyrones can be further exploited to generate molecular diversity using literature reports. Keeping in view the time taken for the completion of reaction in 4-hydroxy (16 h), 4-methoxy (10 h) and 4-chloropyrone (16 h), we can conclude that 1 is more reactive than 19 which in turn is more reactive than 21. In general pyrone 1 is more reactive than pyridone 2 in all the studied reactions.
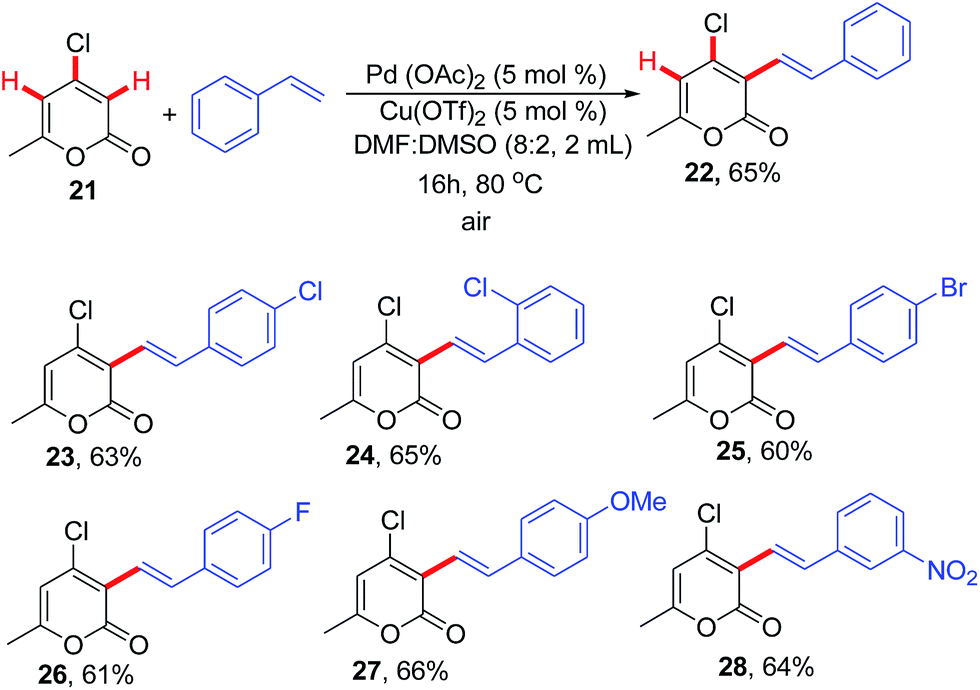 | ||
| Fig. 3 Regio- and chemoselective oxidative cross coupling of 4-chloropyrone and styrenes. | ||
In order to arrive at the regioselectivity of the oxidative C–C bond formation, following plausible mechanism is proposed (Fig. 4). First there is C–H activation where Pd(II) catalyst coordinates with substrate (A) through electrophilic metallation to generate substrate–PdII intermediate B, which is subsequently attacked by substrate alkene through intermolecular Heck type reaction to give an intermediate C. Then, 3-alkenyl-derivative D and Pd(0) would be generated by β-hydride elimination. In order to complete the catalytic cycle, the molecular oxygen under the assistance of Cu(II) would regenerate the catalyst by oxidation of Pd(0) to Pd(II) (Fig. 4). The high regioselectivity can be attributed to the presence of Z groups having coordinating tendency to Pd leading to the formation of stable six membered complex C and also to the inductive effect of the same groups.
 | ||
| Fig. 4 Plausible mechanism for palladium catalysed oxidative C(sp2)–C(sp2) cross coupling. | ||
Conclusions
In summary we have developed palladium catalysed regioselective oxidative C(sp2)–C(sp2) alkenylation process in pyrones and unprotected pyridone. Substrate flexibility and simple reaction procedure are the merits of the reaction. Application of this synthetic strategy to exploit pyrones and pyridones for various diversifications especially generation of fully functionalised derivatives for various biological activities is underway and will be reported in due course of time.Notes and references
- (a) H. J. Jessen and K. Gademann, Nat. Prod. Rep., 2010, 27, 1168–1185 RSC; (b) O. A. Battenberg, M. B. Nodwell and S. A. Sieber, J. Org. Chem., 2011, 76, 6075–6087 CrossRef CAS PubMed; (c) J. M. Dickinson, Nat. Prod. Rep., 1993, 10, 71–98 RSC; (d) R. R. Vardaro, V. DiMarzo, A. Crispino and G. Cimino, Tetrahedron, 1991, 47, 5569–5576 CrossRef CAS; (e) T. Sunazuka, M. Handa, K. Nagai, T. Shirahata, Y. Harigaya, K. Otoguro, I. Kuwajima and S. Omura, Org. Lett., 2002, 4, 367–369 CrossRef CAS PubMed; (f) M. Mineno, Y. Sawai, K. Kanno, N. Sawada and H. Mizufune, Tetrahedron, 2013, 69, 10921–10926 CrossRef CAS PubMed; (g) G. P. McGlacken and I. J. S. Fairlamb, Nat. Prod. Rep., 2005, 22, 369–385 RSC.
- (a) J. A. van Allan, G. A. Reynolds, J. T. Alessi, S. Chie Chang and R. C. Joines, J. Heterocycl. Chem., 1971, 919–922 CrossRef CAS PubMed; (b) A. D. Thomas, Josemin and C. V. Asokan, Tetrahedron, 2004, 60, 5069–5076 CrossRef CAS PubMed; (c) O. Geiseler and J. Podlech, Tetrahedron, 2012, 68, 7280–7287 CrossRef CAS PubMed; (d) I. V. Magedov, M. Manpadi, M. A. Ogasawara, A. S. Dhawan, S. Rogelj, S. V. Slambrouck, W. F. A. Steelant, N. M. Evdokimov, P. Y. Uglinskii, E. M. Elias, E. J. Knee, P. Tongwa, M. Yu Antipin and A. Kornienko, J. Med. Chem., 2008, 51, 2561–2570 CrossRef CAS PubMed.
- (a) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633–639 CrossRef CAS PubMed and refrences cited there in; (b) Y. Wu, J. Wang, F. Mao and F. Y. Kwong, Chem.–Asian J., 2014, 9, 26–47 CrossRef CAS PubMed and references cited therein; (c) O. Basl, J. Bidange, Q. Shuai and C.-J. Li, Adv. Synth. Catal., 2010, 352, 1145–1149 CrossRef PubMed and references cited therein; (d) Y. Yu and G. I. Georg, Chem. Commun., 2013, 49, 3694–3696 RSC; (e) Y. Yu, M. J. Niphakis and G. I. Georg, Org. Lett., 2011, 13, 5932–5935 CrossRef CAS PubMed.
- (a) W. Ma, R. Mei, G. Tenti and L. Ackermann, Chem.–Eur. J., 2014, 20, 15248–15251 CrossRef CAS PubMed; (b) Y. Su, H. Zhou, J. Chen, J. Xu, X. Wu, A. Lin and H. Yao, Org. Lett., 2014, 16, 4884–4887 CrossRef CAS PubMed; (c) M. Bera, A. Modak, T. Patra, A. Maji and D. Maiti, Org. Lett., 2014, 16, 5760–5763 CrossRef CAS PubMed; (d) N. Gigant and J.-E. Bäckvall, Org. Lett., 2014, 16, 4432–4435 CrossRef CAS PubMed.
- (a) M. J. Burns, R. J. Thatcher, R. J. K. Taylor and I. J. S. Fairlamb, Dalton Trans., 2010, 39, 10391–10400 RSC; (b) M.-T. Nolan, J. T. W. Bray, K. Eccles, M. S. Cheung, Z. Lin, S. E. Lawrence, A. C. Whitwood, I. J. S. Fairlamb and G. P. McGlacken, Tetrahedron, 2014, 70, 7120–7127 CrossRef CAS PubMed.
- (a) T. Itahara, Chem. Commun., 1981, 254 RSC; (b) T. Itahara, J. Org. Chem., 1985, 50, 5272–5275 CrossRef CAS; (c) T. Itahara, K. Kawasaki and F. Ouseto, Synthesis, 1984, 236–237 CrossRef CAS.
- Y.-Y. Yu, M. J. Niphakis and G. I. Georg, Org. Lett., 2011, 13, 5932–5935 CrossRef CAS PubMed.
- (a) S. T. McCracken, M. Kaiser, H. I. Boshoff, P. D. W. Boyd and B. R. Copp, Bioorg. Med. Chem., 2012, 20, 1482–1491 CrossRef CAS PubMed; (b) N. Flores, G. Cabrera, I. A. J. Imenez, J. Pinero, A. Gimennez, G. Bourdy, F. Cortes-Selva and I. L. Bazzocchi, Planta Med., 2007, 73, 206–211 CrossRef CAS PubMed; (c) M. G. Pizzolatti, B. G. Mendes, A. Cunha, C. Soldi, A. H. Koga, I. Eger, E. C. Grisard and M. Steindel, Brazilian Journal of Pharmacognosy, 2008, 18, 177–182 CAS; (d) R. Mata, I. Morales, O. Pérez, I. Rivero-Cruz, L. Acevedo, I. Enriquez-Mendoza, R. Bye, S. Franzblau and B. Timmermann, J. Nat. Prod., 2004, 67, 1961–1968 CrossRef CAS PubMed.
- (a) A. K. Kusunuru, S. K. Yousuf, M. Tatina and D. Mukherjee, Eur. J. Org. Chem., 2015, 459–462 CrossRef CAS PubMed; (b) M. Tatina, A. K. Kusunuru, S. K. Yousuf and D. Mukherjee, Org. Biomol. Chem., 2014, 12, 7900–7903 RSC; (c) M. Tatina, A. K. Kusunuru, S. K. Yousuf and D. Mukherjee, Chem. Commun., 2014, 49, 11409–11411 RSC; (d) A. K. Kusunuru, M. Tatina, S. K. Yousuf and D. Mukherjee, Chem. Commun., 2013, 49, 10154–10156 RSC.
- D. Cheng and T. Gallagher, Org. Lett., 2009, 11, 2639–2641 CrossRef CAS PubMed.
Footnotes |
| † IIIM-Publication number: IIIM/1829/2015. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra12631b |
| This journal is © The Royal Society of Chemistry 2015 |