
Imparting magnetic functionality to iron-based MIL-101 via facile Fe3O4 nanoparticle encapsulation: an efficient and recoverable catalyst for aerobic oxidation†
Zhaokui Jina,
Yi Luana,
Mu Yanga,
Jia Tanga,
Jingjing Wanga,
Hongyi Gaoa,
Yunfeng Lub and
Ge Wang*a
aBeijing Key Laboratory of Function Materials for Molecule & Structure Construction, School of Material Science and Engineering, University of Science and Technology Beijing, 30 Xueyuan Road, Beijing, 100083, P. R. China. E-mail: gewang@126.com
bDepartment of Chemical and Biomolecular Engineering, University of California, LosAngeles, CA 90095, USA
First published on 2nd September 2015
Abstract
The development of sustainable, easily synthesizable and highly efficient catalysts is a fundamental goal of catalysis science. In this work, highly superparamagnetic nanoparticles (Fe3O4 NPs) were prepared through a modified co-precipitation method and embedded into a Fe-MIL-101 metal–organic-framework through a facile encapsulation strategy. The catalytic activity of Fe3O4/Fe-MIL-101 was investigated in the aerobic oxidation of alcohols and epoxidation of olefines. High yields (up to quantitative conversion) were observed for both reactions under mild conditions. Furthermore, the magnetic NP/MOF catalysts could be easily recovered and recycled; even after usage in eight consecutive reaction batches, no significant loss of catalytic efficiency was observed. This class of catalysts is promising for green and practical large-scale industrial applications.
1. Introduction
The demand for catalysts suitable for the controlled oxidation of alcohols is rapidly growing, as this reaction is a key step in the industrial synthesis of several fine and specialty products.1,2 Recently, the immobilization of transition-metals on solid supports to synthesize heterogeneous nanocatalysts for aerobic oxidation has become popular in academia and industry. Nanocatalysts have shown good catalytic activity and selectivity and they are easy to handle, recover and recycle.2 Systems based on heterogeneous nanocatalysts and employing readily available oxidants, such as O2 or H2O2, are desirable and have been reported in the literature.3,4 However, they often require toxic or expensive transition metals such as palladium and ruthenium. Only a few examples of aerobic alcohol oxidation promoted by “green metals” have been reported.5,6 Furthermore, the synthesis for existing heterogeneous catalyst carriers, such as zeolite,7,8 alumina9 and hydrotalcite,10,11 is considerably complex and their structure is generally not tuneable. It is therefore important to develop appropriate nanostructures that can support green and inexpensive metal catalysts for highly efficient aerobic alcohol oxidation reaction.Metal–organic frameworks (MOFs) are porous, crystalline materials based on a three-dimensional network of metal ions held in place by multi topical linkers. The development of MOFs has been mostly designed for energy related technologies such as gas storage,12 sensing13 and chemical separation,14 because of their exceptionally large surface area, as well as for a low framework density and tailorable chemistry. Compared to other porous materials, MOFs contain a high density of catalytically active sites, in which the constitutional transition metal has free positions or can easily exchange ligands. For these reasons, the exploitation of MOFs as heterogeneous supports for catalysts looks promising. Several efforts have already been made to use MOFs as supports for heterogeneous catalysts.15–18 In 2008, Ravon and co-workers reported the utilization of Zinc-based MOFs catalysts for the Friedel–Crafts alkylation of aromatic substrates.19 Over the following years, the Garcia group has also presented their studies into the use of aluminum- and copper-based MOFs. Such heterogeneous catalysts were employed in the reduction of carbon–carbon multiple bonds with hydrazine20 and in the aerobic oxidation of variously substituted benzyl alcohols.17
Furthermore, combining MOFs and extrinsic functionalities is also a current research field in catalytic chemistry. The combination of different functionalities into one nanostructure allows the preparation of novel nanocomposite materials with enhanced or new characteristics.21,22 Moreover, the processes for the recovery of nanocatalysts are challenging as conventional filtration becomes extremely difficult as such small scale.23 An efficient and practical route for the recycling of the nanocatalyst is desirable.
In this work, we have developed a novel and highly active heterogeneous Fe3O4/Fe-MIL-101 catalyst. PAA-modified magnetic nanoparticles had activated surface and could act as seeds for the growth of MOFs, subsequently, were encapsulated into polycrystalline MOF aggregates. Magnetic functionalization was thereby achieved in this way without altering their magnetic properties but by forming a strong interaction between the two functional components (Fig. 1). Based on their accessibility, low-toxicity and rich organometallic activity, iron-based MOFs are ideal catalysts for performing organic synthesis. Basolite® F-300 is an MIL-100 iron(III) complex of 1,3,5-benzenetricarboxylate. It has been shown to selectively oxidize cycloalkenes24 and promote the Claisen–Schmidt condensation.25 We prepared the magnetic Fe3O4 dispersion through a facile co-precipitation procedure, which is cheaper and faster when compared to previously reported methods.26,27 In addition, this two-step encapsulation strategy is also faster and more easily scalable, which is a significant improvement when compared to layer-by-layer assembling methods.28 Our approach not only exploits the role of Fe-MOF as an heterogeneous catalyst, but also endows the composite for magnetic recovery via simple encapsulation methods.
 | ||
| Fig. 1 Preparation of functional Fe3O4-NPs/Fe-MOFs materials. | ||
2. Experimental
2.1 Materials
Ferric chloride hexahydrate (FeCl3·6H2O, 97%), ferrous chloride tetrahydrate (FeCl2·4H2O, 98%), terephthalic acid (H2BDC, 98%), polyvinyl pyrrolidone (PVP; Mw = 8000) and 2,2,6,6-tetramethyl-1-piperidinyloxy free radical (TEMPO) were obtained from Alfa Aesar. Polyacrylic acid (PAA; Mw = 1800) was purchased from Sigma-Aldrich. Acetic acid (98%), ammonium hydroxide (NH3·H2O, 25 wt%), anhydrous methanol/ethanol and dimethyl formamide (DMF) were supplied by Beijing Chemical Reagents Company (China). Deionized water was used in all experimental steps that required water.2.2 Catalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, washed with deionized water and ethanol for several times, and dried under vacuum at 40 °C for 12 h. The as-obtained PAA-modified Fe3O4 nanoparticles (0.18 g) were dispersed in 40 mL of DMF containing 0.10 g of PVP. The mixture was sonicated at 300 W for 45 minutes in an ice-water bath until a homogeneous solution was obtained, and left still overnight. The homogenate was filtered and the supernatant served as the stock solution for the following step.
000 rpm, washed with deionized water and ethanol for several times, and dried under vacuum at 40 °C for 12 h. The as-obtained PAA-modified Fe3O4 nanoparticles (0.18 g) were dispersed in 40 mL of DMF containing 0.10 g of PVP. The mixture was sonicated at 300 W for 45 minutes in an ice-water bath until a homogeneous solution was obtained, and left still overnight. The homogenate was filtered and the supernatant served as the stock solution for the following step.2.3 Characterization
The morphology of the as-prepared composites was obtained using scanning electron microscopy (SEM, ZEISS SUPRA55), transmission electron microscopy (TEM, JEM-1200EX, 120 kV) and high resolution transmission electron microscopy (HRTEM, TEI Tecnai F20, 200 kV) equipped with SAED. Powder X-ray diffraction were used to analyze the crystalline phase composition of samples by using a M21X diffractometer (Cu Kα, λ = 1.54056 Å) operated at 40 kV and 200 mA, and the experimental diffraction patterns were collected with step scanning range of 5–80° at room temperature. The nitrogen sorption–desorption isotherms (N2 adsorption) of the composites were used for the analysis of pore structures (Micromeritics ASAP 2420). Fourier transform infrared spectra (FTIR) were recorded using KBr pellet samples with a NICOLET 6700 infrared spectrophotometer. The thermal decomposition behaviours of the samples were analysed by Thermogravimetric Analysis (Netzsch STA449F3 instrument, 10 K min−1 under an air or nitrogen flow). Magnetic measurements for the magnetic composites were carried out with a MPMS-XL superconducting quantum interference device (SQUID) at room temperature. X-ray photoelectron spectroscopy (XPS) analysis was carried out by an Escalab 220i-XL electron spectrometer from VG Scientific using 300 W Al Kα radiation. The binding energies were referenced to the C 1s line at 284.8 eV from adventitious carbon. The catalytic efficiency was calculated by gas chromatography-mass spectrum (GC-MS, Agilent 7890A/5975C with HP-5MS column) and nitrobenzene was used as an internal standard.2.4 Catalytic performance evaluation
2.5 Catalyst recycling
After the reaction was complete, catalyst F1 was recovered by use of an external magnetic field. It was then washed with acetonitrile and methanol to remove any leftover reagents and dried under vacuum at 40 °C. The recyclability of F1 was tested for the aerobic oxidation of alcohols. Each time, F1 was re-used in the presence of a fresh batch of benzyl alcohol, maintaining the same reaction conditions. The mother liquor was analysed by ICP-AES to study the leaching of Fe3+ during the reaction.3. Results and discussion
3.1 Characterization of magnetic MOFs composite
Fe-MIL-101, a class of iron-based MOFs with the molecular formula Fe3O(BDC)3, has a zeotype topology. It comprises two kinds of cages with free internal diameters of 29 and 34 Å respectively. These cages are accessible through microporous windows of ∼12–16 Å.31,32 Fe-MIL-101 was chosen in this work for the incorporation of Fe3O4 NPs because it possesses a high density of accessible catalytic centres (FeIII) and relatively high thermal stability. Furthermore, magnetic nanoparticles can be successfully incorporated inside MOFs with carboxylic-acid-group ligands. An appropriate surface modification of magnetic Fe3O4 NPs employing polyacrylic acid was prepared with a facile co-precipitation method and used to prevent the aggregation of Fe3O4 NPs, leading to stable and dispersed nanoparticles incorporated in the MOFs structure. The additional PVP promotes continuous formation of fresh surfaces during the growing of MOFs crystal; PAA-modified Fe3O4 NPs can successfully adhere to the growing MOFs crystal without breaking the crystal structure of MIL-101.26The morphology and structure of the as-prepared Fe3O4@Fe-MIL-101 (F1) nanocomposite were characterized by SEM and TEM. The composite shows a concave octahedral morphology with an average diameter of 300 nm, which corresponds with the MOFs matrix (Fig. 2a). Most octahedrons have a smooth surface and a few particles adhere to each other (Fig. 2b). TEM images clearly display the regular octahedral crystalline structures of MOFs and Fe3O4 NPs encapsulated therein (black in colour Fig. 2c). HRTEM image (Fig. 2d) clearly shows that most of the Fe3O4 NPs are dispersed inside the octahedron; the average size of the Fe3O4 NPs is about 15 nm, indicating that the encapsulation strategy was successful. The electron diffraction (SAED) pattern, visible in the selected area of F1 (insert in Fig. 2d), confirms the crystalline feature of Fe3O4 NPs.33
 | ||
| Fig. 2 SEM and TEM images of Fe3O4/Fe-MIL-101 nanocomposite F1: (a and b) SEM; (c and d) TEM. The insert in (d) is the corresponding electron diffraction (SAED) pattern of the selected area. | ||
The crystal structure, surface chemical composition and porosity of F1 were investigated by XRD, XPS, and nitrogen adsorption–desorption isotherm measurement, respectively. The composite F1 exhibited identical diffraction patterns to the intrinsic MIL-101, indicating that this encapsulation maintained the intact crystalline form of the composite (as shown in Fig. 3). Apparently expected feature of Fe3O4 is absent, which might be due to the low concentration and/or small sizes of Fe3O4 domains.21,26 The XPS spectra of F1 were measured in order to further understand the surface composition and are reported in Fig. 4a and in the ESI (in Fig. S2†). The main peaks are C 1s, O 1s and Fe 2p; the binding energies of F1 for Fe 2p, are found to be 711.5 eV and 725.2 eV and can be attributed to the Fe 2p3/2 and Fe 2p1/2, respectively (Fig. 3c). The separation Δ = 2p1/2 − 2p3/2 = 13.5 eV is similar to those reported for Fe2O3.34 The peaks may therefore belong mainly to the FeIII centres of the Fe-MIL-101 matrix. The FeII signal is absent from this spectrum, indicating that no encapsulated Fe3O4 are detected by XPS measurement. This further supports that the magnetic Fe3O4 NPs are encapsulated within the MOFs matrix. Even after eight consecutive reuses in synthetic applications (see below, in Section 3.3, for details), the XPS spectrum did not change significantly, confirming the high chemical stability of the composite Fe3O4-NPs/MOFs as a heterogeneous catalyst. Fig. 4b shows that there is a steep increase in nitrogen uptake at low relative pressure in the nitrogen-sorption isotherms, suggesting that catalyst F1 possesses both micropores and mesopores. In the pore size distribution curve (Fig. 4a), pore diameter centres at 1.3 and 5.3 nm are visible. The surface area calculated by a BET method is 1230 m2 g−1, which is lower than that of MIL-101 obtained by the solvothermal method (1458 m2 g−1). This is most likely due to the non-porous Fe3O4 particles and the persistence of PVP in the composites. However, it is still considerably higher than those of other magnetic porous catalysts,25,35 implying our composite is superior as a heterogenous catalyst for high-efficiency catalysis.
 | ||
| Fig. 3 XRD patterns of (a) the simulated from the crystallographic data of MIL-101(Fe) and (b) the experimentally acquired crystallographic data of catalyst F1. | ||
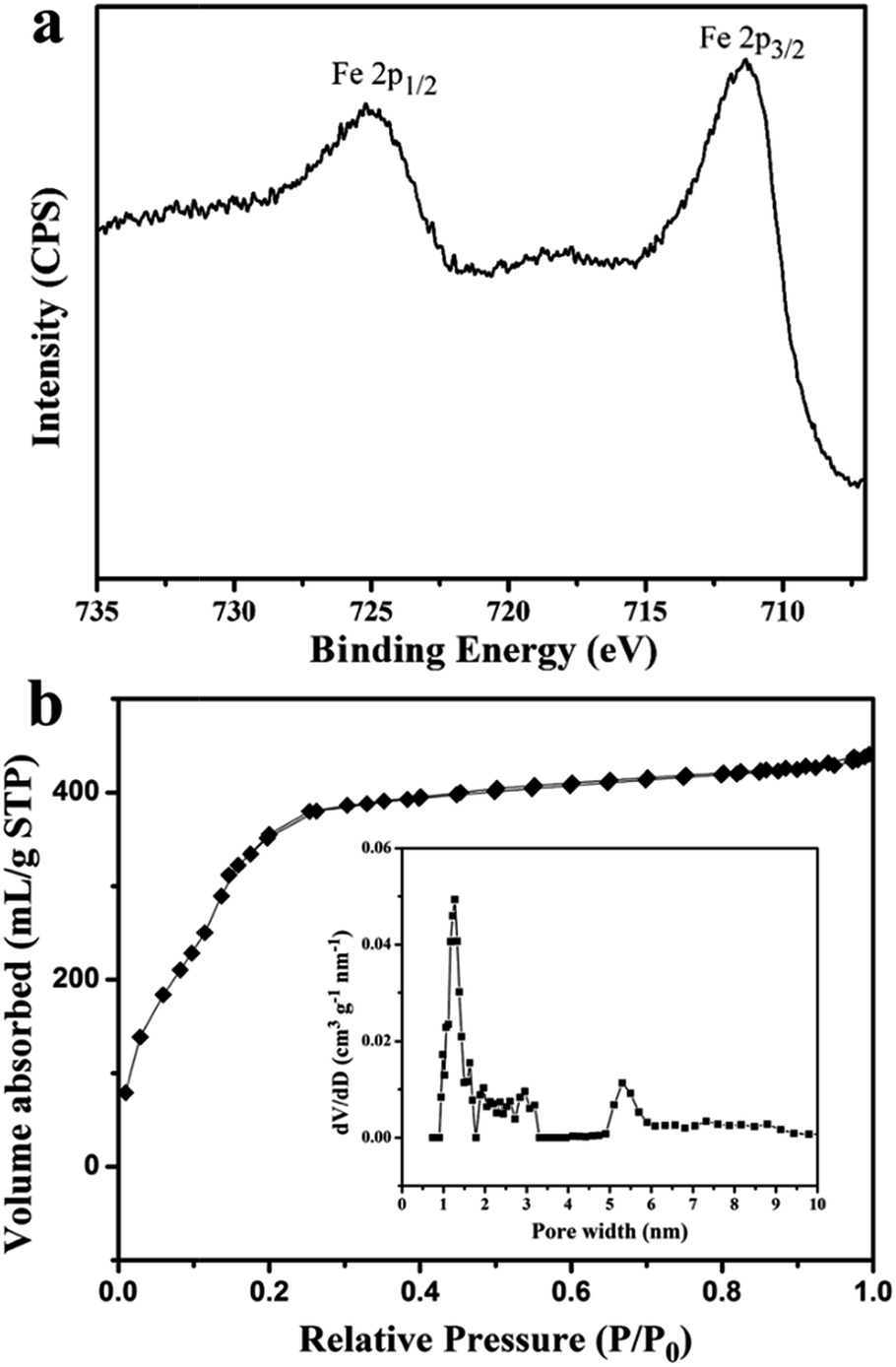 | ||
| Fig. 4 (a) High resolution Fe 2p spectra of F1 (b) nitrogen adsorption–desorption isotherms of F1, and its corresponding pore size distribution curve (insert). | ||
The magnetic properties and the thermostability of F1 were investigated by vibrating sample magnetometry (VSM) and thermogravimetric analysis (TGA), respectively. The magnetic susceptibility μ was measured at room temperature using vibrating sample magnetometry (VSM) in the field range from −10 to +10 kOe. The magnetic hysteresis curve for F1 (Fig. 5) displays no remanence or coercivity at room temperature, showing the superparamagnetic behaviour of the hybrid material. The saturation magnetization value of F1 is 2.1 emu g−1, which suggests a low magnetite content (∼3.40 wt%) in the prepared composites, when compared to pure Fe3O4 NPs (μ = 59 emu g−1), which is close to the feed ratio (3.84 wt%). Synthetically, the magnetic properties of the composite are important for its recovery from the reaction mixture. The composites can in fact be easily separated from the solution by mean of approaching an external magnet to the reaction mixture (Fig. 5, inset). Similarly to Fe-MOFs, F1 has a relatively high decomposition temperature of about 400 °C; the amount of decomposition observed for F1 at such temperature is marginally larger than that observed for intrinsic Fe-MOFs and may be attributable to the decomposition of the PAA and PVP chains.
 | ||
| Fig. 5 VSM curves of F1 at room temperature. Inset: photograph of the recovery of the composite from an aqueous suspension. The picture shows the visible change brought about by placing a neodymium magnet next to the vial containing the suspension. The Fe3O4-NP–MOFs migrate towards the magnet and can be easily recovered after the solution is removed. | ||
3.2 Catalytic activity of Fe3O4-NPs/MOFs composite
The prepared composite materials of Fe3O4-NPs/MOFs were tested for catalytic activity. Two systems were chosen for their synthetic importance: (1) the aerobic oxidation of alcohols and (2) the aerobic epoxidation of olefins.1,2 The application of Fe3O4-NPs/MOFs as a heterogeneous catalyst proved very efficient in both reactions; high yields and very high selectivities were obtained.
|
||||||
|---|---|---|---|---|---|---|
| Entry | TEMPO (eq.) | KNO2 (eq.) | Catalyst | Solvent | Conv.a (%) | Select.a (%) |
| a Conversion (%) and selectivity (%) were determined by GC-MS using nitrobenzene as the internal standard.b 56 °C.c 40 °C. | ||||||
| 1 | 0.5 | 0.2 | — | CH3CN | Trace | — |
| 2 | 0.5 | 0.2 | Fe3O4 | CH3CN | Trace | — |
| 3 | 0.5 | 0.2 | FeCl3 | CH3CN | 100 | >99 |
| 4 | 0.5 | 0.2 | F1 | CH3CN | 100 | >99 |
| 5b | 0.5 | 0.2 | F1 | Acetone | 79 | >99 |
| 6c | 0.5 | 0.2 | F1 | CH2Cl2 | 57 | >99 |
| 7 | 0.5 | 0.2 | F1 | THF | 18 | >99 |
| 8 | 0.5 | 0.2 | F1 | EtOH | 6 | >99 |
| 9 | — | 0.2 | F1 | CH3CN | Trace | >99 |
| 10 | 0.5 | — | F1 | CH3CN | 73 | >99 |
| 11 | 0.25 | 0.2 | F1 | CH3CN | 67 | >99 |
| 12 | 0.5 | 0.2 | F2 | CH3CN | 47 | >99 |

As a general trend, the percent-conversion of the reagent increases as the polarity of solvent decreases, suggesting that aprotic solvents with higher dipole moments and dielectric constants are beneficial to the process. Presumably, aprotic polar solvents improve the dispersibility of the heterogeneous catalyst, increasing the diffusion rate and resulting in more interactions between the substrate and the catalyst. Varying the amount of TEMPO present in the solution greatly affected the reaction; no benzaldehyde formation was observed in the absence of TEMPO and only a lower yield was measured in the presence of a reduced amount of it (Table 1, entry 9 and 11, respectively). Unexpectedly, KNO2 was found to affect the reaction only marginally; comparable efficiencies were achieved with lower amounts than those found in the literature for iron–TEMPO–KNO2 system36 (Table 1, entries 10 and 4). Therefore, the optimized catalytic procedure involves stirring for 14 h at 75 °C the following reaction mixture: 0.1 eq. MOF-based catalyst, 0.5 eq. TEMPO as co-catalyst, 0.2 eq. KNO2 as an additive per equivalent of substrate, and molecular oxygen as an oxidant in acetonitrile.
An analogue composite Fe3O4/Fe-MIL-88B (named F2) was also prepared. The morphology of F2 involved a bipyramidal hexagonal prism with a small BET value and narrow pore diameter. The relevant synthetic methodology and the full characterization is provided in part 1 of the ESI.† While F1 and F2 share the same composition and a similar topology, F1 shows better oxidation activities than F2 (Table 1, entry 12). This is presumably due to the larger aperture in the structure and the higher specific surface area of F1. This result suggests that suitable pore structure played an important role during heterogeneous catalysis. Our Fe3O4/Fe-MIL-101 system showed improved catalytic performance than several literature reported systems. The turnover number (TON) of F1 is calculated to be 9.9, while the reaction is performed under a mild condition. Molecular sieves was used as catalyst and only a TON of 1.7 was achieved in toluene solvent. Combined metal oxide gave similar TONs, however, in an elevated reaction temperature and extended reaction time (Table S1,† entries 2 and 3). The immobilization of Cu(II) on molecular sieves supports also gave a compromised TON (Table S1,† entry 4). The TON was slightly improved in an uncommon and expensive solvent (Table S1,† entry 5). The iron-based heterogeneous catalyst for aerobic oxidation of alcohols is rare. The iron exchanged MPA gave a TON of 21, however, only 35% conversion was achieved after 20 h. Also, Cu-based MOFs failed to provide high TON due to the high catalyst usage and low catalyst efficiency (Table S1,† entry 7). In summary, our catalyst involving magnetic functionality originated from facile Fe3O4 nanoparticles encapsulation demonstrated good catalytic efficiency as novel heterogeneous catalyst for aerobic oxidations under mild reaction condition.
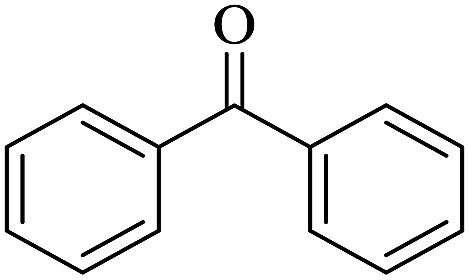
In order to investigate the applicability of the magnetic catalyst F1, the reaction was tested on a wide variety of primary and secondary alcohols. The results are summarized in Table 2. Various aromatic alcohols bearing electron-donating or electron-withdrawing functional groups were tested (Table 2, entries 1–4). As expected, the aryl alcohols with electron-donating substituents like methyl and methoxy groups gave outstanding yields (99%) to the corresponding aromatic aldehydes with >99% selectivity. For the aryl alcohols with electron-withdrawing substituents instead, like a fluorine atom or a nitro group, slightly lower yields were obtained. This is most likely due to the lower electron density present at the benzylic position. It has to be noted that the heterocyclic substrate, 2-pyridine methanol, can be converted into the corresponding aldehyde with excellent yield (Table 2, entries 5). In addition, allylic alcohols, such as 3-methyl-2-buten-1-ol and cinnamyl alcohol, were neatly converted into the corresponding aldehydes in moderate yields (Table 2, entries 6 and 7); no C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond reactivity was observed. On the other hand, secondary alcohols like 1-phenylethanol and cyclohexanol are inert to be oxidized to the corresponding ketones (Table 2, entries 8 and 9). Lastly, the aerobic oxidation of benzhydrol (Table 2, entry 10) did not proceed under the optimised condition, which is probably due to steric interactions and low reactivity.
C double bond reactivity was observed. On the other hand, secondary alcohols like 1-phenylethanol and cyclohexanol are inert to be oxidized to the corresponding ketones (Table 2, entries 8 and 9). Lastly, the aerobic oxidation of benzhydrol (Table 2, entry 10) did not proceed under the optimised condition, which is probably due to steric interactions and low reactivity.
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Conv.b (%) | Select.b (%) |
| a Reaction conditions: 1.0 mmol substrate in 5.0 mL CH3CN was heated at 75 °C with F1 (0.1 mmol), TEMPO (0.5 mmol), KNO2 (0.2 mmol) and 1.0 atm O2 ballon for 14 h.b Conversion (%) and selectivity (%) were determined by GC-MS using nitrobenzene as the internal standard. | ||||
| 1 | 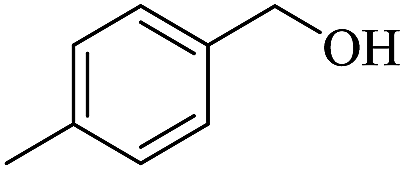 |
 |
99 | >99 |
| 2 | 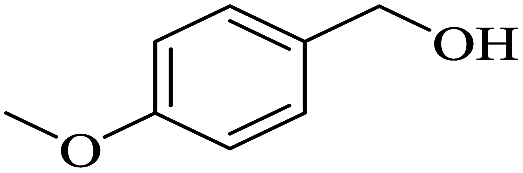 |
 |
99 | >99 |
| 3 |  |
 |
93 | >99 |
| 4 |  |
 |
82 | >99 |
| 5 |  |
 |
99 | >99 |
| 6 |  |
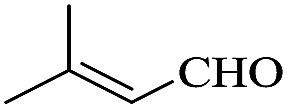 |
60 | >99 |
| 7 |  |
 |
65 | >99 |
| 8 |  |
 |
15 | >99 |
| 9 | 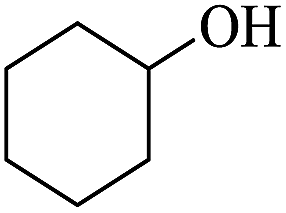 |
 |
9 | >99 |
| 10 |  |
 |
— | — |
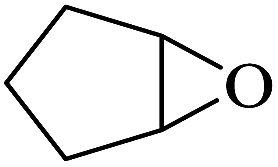
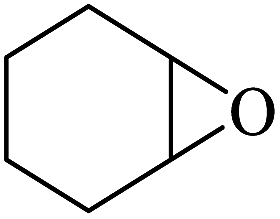
 :54, while trans-stilbene was oxidized to the specific trans-stilbene oxide (entries 6 and 7).40–42 Due to the installation of four bulky tert-butyl groups, cis-3,3′,5,5′-tetra(tert-butyl)stilbene became significantly larger (13.7 × 7.8 Å) than the available pore size of F1 and gives a much lower yield (Table 3, entry 8). This suggests that the aerobic epoxidation catalysed by the MOFs composite F1 has size and shape selectivity.
:54, while trans-stilbene was oxidized to the specific trans-stilbene oxide (entries 6 and 7).40–42 Due to the installation of four bulky tert-butyl groups, cis-3,3′,5,5′-tetra(tert-butyl)stilbene became significantly larger (13.7 × 7.8 Å) than the available pore size of F1 and gives a much lower yield (Table 3, entry 8). This suggests that the aerobic epoxidation catalysed by the MOFs composite F1 has size and shape selectivity.
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Conv.b (%) | Sel.b (%) |
| a Reaction conditions: 1.0 mmol substrate in 5.0 mL CH3CN was heated at 25 °C with F1 (0.027 mmol), (CH3)3CCHO (2.0 mmol) and 1.0 atm O2 balloon for 4 h.b Conversion (%) and selectivity (%) were determined by GC−MS through an internal standard method.c Determined by 1H-NMR.d The main product was benzaldehyde. | ||||
| 1 |  |
 |
95 | 79 |
| 2 |  |
 |
96 | 81 |
| 3 |  |
 |
98 | 95 |
| 4 |  |
 |
98 | 96 |
| 5 |  |
 |
98 | 96 |
| 6 |  |
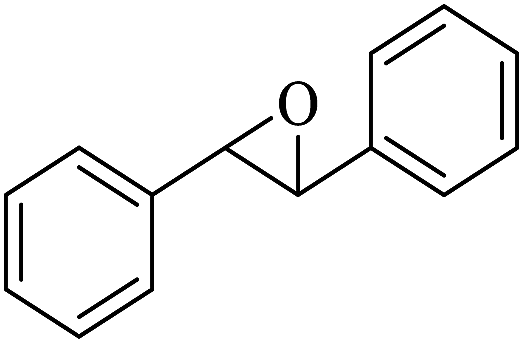 |
67 | 99 |
| 7 |  |
 |
60 | 99 |
| 8 |  |
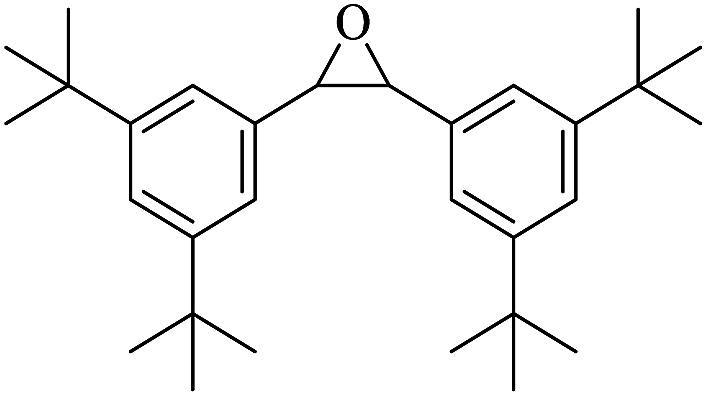 |
15 | 99c |
| 9 |  |
 |
11 | —d |

The epoxidation of styrene showed a low yield of the desired epoxide, due to the formation of major amounts of byproduct. The dominant reaction involved in fact the formation of benzaldehyde as the major product. The kinetic product of epoxidation is unstable and can be further oxidized to the thermodynamically more stable benzaldehyde (Table 3, entry 9).43,44
Overall, the catalytic system proved to have several synthetic advantages over existing systems, such as NHPI/Fe(BTC),24 and Mn(III)/MCM-41.45 Fe3O4-NP/MOF-based catalysts are able to efficiently perform the epoxidation of olefins using a considerably lower catalyst loading, at room temperature and in a shorter time. Further work is needed to design similar catalysts for the oxidation of larger and less reactive substrates.
3.3 Re-usability of the Fe3O4-NP/MOF catalyst
In order to further evaluate the potential for industrial applications of the Fe3O4-NP/MOF F1, we decided to test the recyclability of the composite material for use with successive batches of fresh benzyl alcohol. The reaction was performed using optimized conditions (Table 1, Entry 3). Upon completion of the reaction, the catalyst was recovered using an external magnet (Fig. 5, inset). The as-recovered composite was then used as the catalyst for the oxidation of a fresh batch of benzyl alcohol.This procedure was repeated for a total of eight times using the same composite material and the yield of benzaldehyde obtained from each batch was quantified. Although it progressively diminished, the yield of the reaction remained good (88%) even after eight re-uses of the catalyst (Fig. 6). The SEM, TEM, XRD, VSM and XPS results (reported in the ESI, in Fig. S4–S8†) show that the structure, composition and magnetic property of the catalyst were unchanged even after eight runs. The BET surface area of re-used catalyst decreased slightly from 1230 to 973 m2 g−1 (ESI, Fig. S9†), which may be due to partial blockage of the channels by insoluble residue.
 | ||
| Fig. 6 Recycling of F1 catalyst in the aerobic oxidation of benzyl alcohol. | ||
3.4 Heterogeneity of the Fe3O4-NP/MOF catalyst
Finally, we decided to run a leaching test to investigate whether this catalytic system is truly heterogeneous or whether catalysis is promoted homogeneously by an amount of iron species leaching into the solution.The reaction was initially set up using optimized conditions (Table 1, entry 3), in the presence of an eight-times re-used batch of the catalyst. After six hours of reaction time, catalyst F1 was removed by hot filtration and the solution was left to stir for eight additional hours. Fig. 7 shows the conversion of benzyl alcohol as a function of time with a batch of eight-times re-used catalyst (black curve) and how no further reagent consumption was detected after the catalyst was removed from the mixture (red curve).
The supernatant was analysed by inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis. A concentration of 1.6 ppm of Fe3+ ions was found, revealing that only 0.08% of the iron present in the catalyst had leached into the solution. These results indicated that the magnetic MOFs composite is stable under the applied reaction conditions. The catalytic process is therefore truly heterogeneous.
In summary, F1 is a novel Fe3O4-NP/MOF-based noble-metal-free heterogeneous catalyst, which is capable of high catalytic efficiency towards aerobic alcohol oxidation and olefin epoxidation. The good thermal properties of crystalline MOF composite materials provide a reliable, stable structure that allows high catalyst reusability.
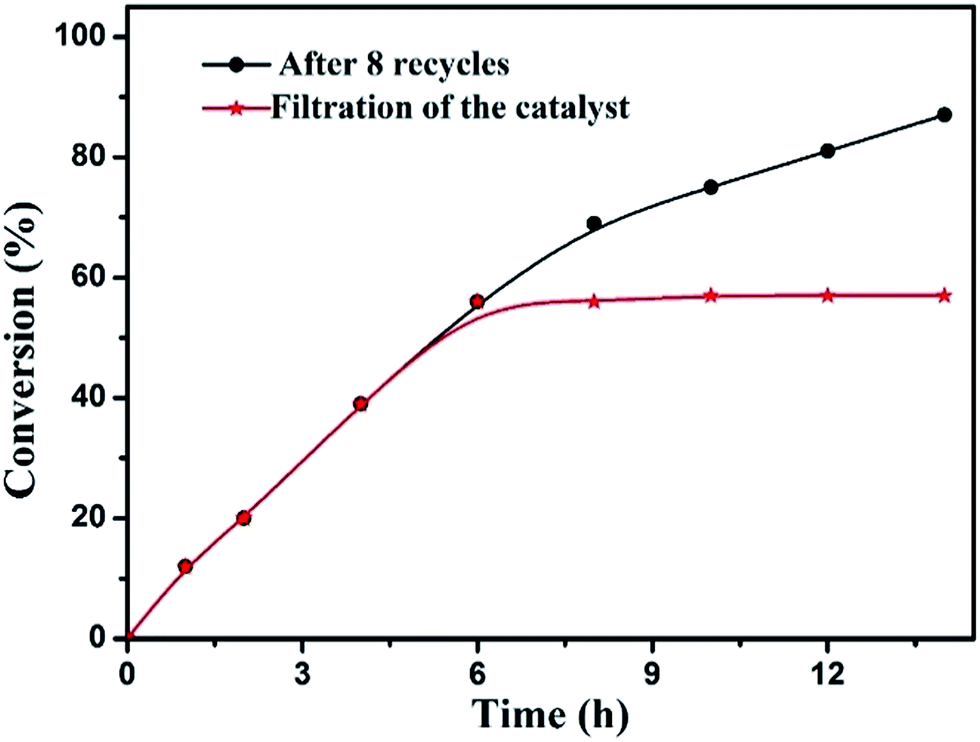 | ||
| Fig. 7 Leaching test for catalyst F1 in the aerobic oxidation of benzyl alcohol. | ||
4. Conclusions
A series of novel magnetic Fe3O4-NP/MOF-based composite catalysts was prepared by an efficient encapsulation method associated with different Fe-MOFs. A facile co-precipitation method for highly dispersed Fe3O4 NPs was modified and employed. Using such techniques, magnetically separable catalysts were obtained.The Fe3O4/Fe-MIL-101 composite catalyst F1 demonstrated remarkable efficiency for the aerobic oxidation of alcohols and the epoxidation of olefins. A wide range of acrylic and allylic alcohols was successfully oxidised taking advantage of the catalyst's large surface area, appropriate pore size and abundance of accessible active sites. The Fe3O4-NP/Fe-MIL-101 composite catalyst F1 also showed catalytic activity for the reaction of the epoxidation of olefins. Interestingly, size and shape selectivity were observed.
Furthermore, re-usability of the catalyst in the process of oxidation of alcohols was proven for at least eight times, with only marginal loss in efficiency. Finally, a leaching test demonstrated that this catalytic system is heterogeneous. In conclusion, functional integration of inexpensive magnetic nanoparticles with ferric nanocomposite materials makes Fe3O4-NP/Fe-MOF a sustainable, environmentally friendly and economic catalyst for oxidation processes.
Acknowledgements
This work is supported by the National High Technology Research and Development Program of China (863 program) (No. 2013AA031702) and Co-building Special Project of Beijing Municipal Education Commission for financial support. We thank Prof. Wenjun Dong and Dr Mark Buck for helpful discussions and English editing.Notes and references
- M. Hudlicky, Oxidation in Organic Chemistry, ACS Monograph Series no. 186, American Chemical Society, Washington, DC, 1990 Search PubMed.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS PubMed.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977–1986 RSC.
- K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2002, 41, 4538–4551 CrossRef CAS.
- I. E. Marko, A. Gautier, R. L. Dumeunier, K. Doda, F. Philippart, S. M. Brown and C. J. Urch, Angew. Chem., Int. Ed., 2004, 43, 1588–1591 CrossRef CAS PubMed.
- Y. Tao, H. Kanoh, L. Abrams and K. Kaneko, Chem. Rev., 2006, 106, 896–910 CrossRef CAS PubMed.
- B.-Z. Zhan, M. A. White, T.-K. Sham, J. A. Pincock, R. J. Doucet, K. V. R. Rao, K. N. Robertson and T. S. Cameron, J. Am. Chem. Soc., 2003, 125, 2195–2199 CrossRef CAS PubMed.
- S. F. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593–8596 CrossRef CAS PubMed.
- K. Ebitani, H.-B. Ji, T. Mizugaki and K. Kaneda, J. Mol. Catal. A: Chem., 2004, 212, 161–170 CrossRef CAS PubMed.
- T. Nishimura, N. Kakiuchi, M. Inoue and S. Uemura, Chem. Commun., 2000, 1245–1246 RSC.
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Adv. Synth. Catal., 2010, 352, 711–717 CrossRef CAS PubMed.
- J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, ACS Catal., 2011, 1, 48–53 CrossRef CAS.
- Y. Zhang and J. Y. Ying, ACS Catal., 2015, 5, 2681–2691 CrossRef CAS.
- U. Ravon, M. E. Domine, C. Gaudillere, A. Desmartin-Chomel and D. Farrusseng, New J. Chem., 2008, 32, 937–940 RSC.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Adv. Synth. Catal., 2009, 351, 2271–2276 CrossRef CAS PubMed.
- W. Zhang, G. Lu, C. Cui, Y. Liu, S. Li, W. Yan, C. Xing, Y. R. Chi, Y. Yang and F. Huo, Adv. Mater., 2014, 26, 4056–4060 CrossRef CAS PubMed.
- X. Gu, Z.-H. Lu, H.-L. Jiang, T. Akita and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11822–11825 CrossRef CAS PubMed.
- T. Cheng, D. Zhang, H. Li and G. Liu, Green Chem., 2014, 16, 3401–3427 RSC.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, J. Catal., 2012, 289, 259–265 CrossRef CAS PubMed.
- F. Ke, L. Qiu and J. Zhu, Nanoscale, 2014, 6, 1596–1601 RSC.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS PubMed.
- I. Imaz, J. Hernando, D. R. Molina and D. Maspoch, Angew. Chem., Int. Ed., 2009, 48, 2325–2329 CrossRef CAS PubMed.
- F. Ke, L. G. Qiu, Y. P. Yuan, X. Jiang and J. F. Zhu, J. Mater. Chem., 2012, 22, 9497–9500 RSC.
- X. Q. Liu, Y. P. Guan, Z. Y. Ma and H. Z. Liu, Langmuir, 2004, 20, 10278–10283 CrossRef CAS PubMed.
- J. Tang, M. Yang, M. Yang, J. Wang, W. Dong and G. Wang, New J. Chem., 2015, 39, 4919–4923 RSC.
- Y. K. Hwang, D.-Y. Hong, J.-S. Chang, H. Seo, M. Yoon, J. Kim, S. H. Jhung, C. Serre and G. Férey, Appl. Catal., A, 2009, 358, 249–253 CrossRef CAS PubMed.
- I. Y. Skobelev, A. B. Sorokin, K. A. Kovalenko, V. P. Fedin and O. A. Kholdeeva, J. Catal., 2013, 298, 61–69 CrossRef CAS PubMed.
- J. Mondal, T. Sen and A. Bhaumik, Dalton Trans., 2012, 41, 6173–6181 RSC.
- M. Descostes, F. Mercier, N. Thromat, C. Beaucaire and M. Gautier-Soyer, Appl. Surf. Sci., 2000, 165, 288–302 CrossRef CAS.
- Y. Xu, H. Zhang, X. Duan and Y. Ding, Mater. Chem. Phys., 2009, 114, 795–801 CrossRef CAS PubMed.
- A. Dijksman, A. Mmarino-Gonzalez, A. Mairata, I. Payeras, I. W. C. E. Arends and R. A. Sheldon, J. Am. Chem. Soc., 2001, 123, 6826–6833 CrossRef CAS PubMed.
- Q. H. Xia, H. Q. Ge, C. P. Ye, Z. M. Liu and K. X. Su, Chem. Rev., 2005, 105, 1603–1622 CrossRef CAS PubMed.
- N. V. Maksimchuk, K. A. Kovalenko, V. P. Fedin and O. A. Kholdeeav, Chem. Commun., 2012, 6812–6814 RSC.
- J. Sun, G. Yu, Q. Huo, Q. Kan and J. Guan, RSC Adv., 2014, 4, 38048–38054 RSC.
- K. Hasan, N. Brown and C. M. Kozak, Green Chem., 2011, 13, 1230–1237 RSC.
- W. Adam, K. J. Roschmann, C. R. Saha-Möller and D. Seebach, J. Am. Chem. Soc., 2002, 124, 5068–5073 CrossRef CAS PubMed.
- Y. Qi, Y. Luan, J. Yu, X. Peng and G. Wang, Chem.–Eur. J., 2015, 21, 1589–1597 CrossRef CAS PubMed.
- C. Q. Chen, J. Qu, C. Y. Cao, F. Niu and W. G. Song, J. Mater. Chem., 2011, 21, 5774–5779 RSC.
- D. H. Tang, W. T. Zhang, Y. L. Zhang, Z. A. Qiao, Y. L. Liu and Q. S. J. Huo, J. Colloid Interface Sci., 2011, 356, 262–266 CrossRef CAS PubMed.
- Y. Zhang, J. Zhao, L. He, D. Zhao and S. Zhang, Microporous Mesoporous Mater., 2006, 94, 159–165 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of F2; SEM, TEM, N2 sorption/desorption isotherms, XPS, VSM of F1. See DOI: 10.1039/c5ra17180f |
| This journal is © The Royal Society of Chemistry 2015 |