
A mild CuBr–NMO oxidative system for the coupling of anilines leading to aromatic azo compounds†
Abstract
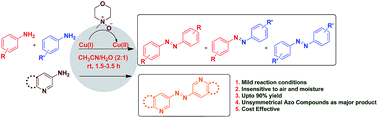
An efficient, mild and cost-effective method has been developed utilizing CuBr with N-methylmorpholine N-oxide (NMO/NMMO) for the oxidative coupling of anilines to access symmetrical and unsymmetrical azo compounds in high yield. The reactivity was found to be governed by electronic and steric factors of anilines.
 Please wait while we load your content...
Please wait while we load your content...

