
Development and characterization of single step self-assembled lipid polymer hybrid nanoparticles for effective delivery of methotrexate†
Abstract
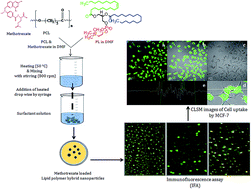
The present study was designed to develop methotrexate (MTX) loaded lipid polymer hybrid nanoparticles (LPHNPs) for spatial and controlled delivery of this drug. LPHNPs were formulated by single step self-assembled nano-precipitation method. The effect of variables such as surfactants, varying surfactant concentration, phospholipids and lipid–polymer ratio on particle size and drug entrapment efficiencies were systematically assessed to optimize LPHNPs. The formulated LPHNPs were found in nanometric size range (150–300) with spherical shape. The entrapment efficiency of developed formulations was calculated between 80 and 90%. The entrapment of drug into nanoparticles was further validated by FTIR and XRD analysis. In vitro drug release study showed slow and sustained drug release i.e. more than 80% at the end of 7th day. The drug release followed Korsmeyer–Peppas model showing Fickian diffusion. The in vitro cytotoxicity of optimized formulations was assessed in MCF-7 cells by MTT, immunofluorescence assay (IFA) as well as confocal laser scanning microscopy (CLSM). The result obtained with in vitro cell line studies suggested that LPHNP is a prominent delivery vehicle for MTX in breast cancers.
 Please wait while we load your content...
Please wait while we load your content...

