DOI:
10.1039/C5RA12131K
(Paper)
RSC Adv., 2015,
5, 74692-74704
An in situ prepared photo-luminescent transparent biocompatible hyperbranched epoxy/carbon dot nanocomposite
Received
23rd June 2015
, Accepted 26th August 2015
First published on 26th August 2015
Abstract
A photo-luminescent transparent biocompatible hyperbranched epoxy/carbon dot nanocomposite was prepared by incorporation of carbon dots during formation of hyperbranched epoxy resin. The prepared nanocomposite was characterized by FTIR, NMR and TEM analyses. The poly(amido-amine) cured nanocomposite exhibited high tensile strength (62.5 MPa), high elongation at break (45%), good thermal stability (291 °C), high transparency and excellent wavelength dependent photoluminescence behavior along with biocompatibility with skin cells. The performance of this nanocomposite was also compared with the pristine hyperbranched epoxy as well as hyperbranched epoxy/carbon dot nanocomposite obtained through an ex situ solution technique. The study revealed that the in situ prepared nanocomposite possessed superior mechanical, optical and biocompatible properties compared to pristine epoxy as well as the ex situ prepared nanocomposite. Thus, the study will significantly contribute to the field of high performance transparent fluorescent polymeric materials used in optoelectronics. Good viability, spreading and proliferation of skin fibroblasts and keratinocyte cells on the nanocomposite suggest it is also a highly potential material for bio-sealant application.
1. Introduction
In the last two decades intensive research has been focused on quantum dots (semiconductor nanocrystals) because of their promising optical and electronic properties.1–3 Recently polymer/quantum dot nanocomposites have captured tremendous interest to achieve high transparency and high luminescence solid films for advanced optoelectronics.2,4–6 The polymer matrix provides mechanical and chemical stability as well as helps in dispersion of the quantum dots by preventing their agglomeration. However, because of the organophobic surface of semiconductor quantum dots, good dispersion in a polymer matrix is a key challenge to obtain highly transparent photoluminescent films even by an in situ polymerization technique.7,8 In case of semiconductor quantum dot based nanocomposite biocompatibility of the material is also a major issue. In this respect, carbon quantum dot or carbon dot (size 2–10 nm) is a new class of fluorescent material as an attractive alternate to traditional semiconductor nanocrystals due to superior properties like resistance to photobleaching, chemical inertness, no optical blinking, easy functionalizability, non toxicity or biocompatibility and water solubility of the former.9–13 The presence of large number of oxygenous functional groups on its surface provides excellent dispersion in many polar solvents as well as in polymer matrices. Simple synthetic protocols and use of easily available carbon precursors make it very cost-effective. Recently research has been started on carbon dot based light emitting polymer nanocomposites for different optoelectronics including light emitting device (LED). Zhou et al. synthesized polysaccharide based amphibious fluorescent carbon dot and incorporated it into poly(methyl methacrylate) and poly(vinyl alcohol) matrices to obtain light emitting polymer nanocomposites.14 Hao et al. incorporated carbon dot into poly(ethylene glycol) to produce poly(ethylene glycol)/carbon dot nanocomposite film for tunable blue-red light emission.15 Kwon et al. fabricated a freestanding luminescent film from poly(methyl methacrylate) and nitrogen-rich carbon dot for white light emitting device.16 However, in all these cases nanocomposites are prepared by ex situ technique. Further the performance including mechanical properties, thermal stability, etc. was also not reported by these investigations. On the contrary, carbon dot has large number of polar functionalities and thus provides an opportunity to be used as a nanomaterial for in situ polymer nanocomposite. This not only helps in uniform and stable dispersion of carbon dot but it also aids to the performance of the nanocomposite in a significant manner.
On the other hand attributes of high mechanical properties, high thermal stability, excellent adhesive strength, good weather and chemical resistance, high transparency and others endow a unique position to epoxy resins in the domain of optoelectronics.17–20 Last decade, researchers have trusted their interests on luminescent epoxy nanocomposites by incorporating different fluorescent nanomaterials for making advanced optoelectronic devices. Zou et al. reported a transparent and luminescent epoxy nanocomposite with CdSe quantum dots.21 Yang et al. reported transparent and light-emitting epoxy nanocomposites containing ZnO quantum dots as encapsulating materials for solid state lighting.22 However, in all afore mentioned cases, mainly toxic semiconductor quantum dots were used and no report on performance of the materials was noticed. However, the improvement of performance and luminescence properties are limited for advanced optoelectronics in all such cases including our recent report,23 as they are ex situ nanocomposites. Further, carbon dot based polymer or polymer nanocomposite not only offers high performance fluorescent material, it also introduces biocompatibility to the system which is helpful for bio-imaging application.24 However, in case of carbon dot based biocompatible in situ epoxy nanocomposite is the best suitable material for the bio-sealant application in surgical wound healing along with bio-imaging.
Thus, in the present report a high performance photo-luminescent transparent hyperbranched epoxy nanocomposite with biocompatibility attribute was prepared where carbon dot was incorporated during preparation of the epoxy resin. The hyperbranched structure of such epoxy added special interest due to its easy synthetic accessibility and processibility, high solubility, low viscosity, high reactivity and large number of end functional groups.19,20,25
2. Experimental section
2.1. Materials
Carbon dot used in this study was prepared by heating of banana juice at 150 °C as reported earlier.26 Pentaerythritol (Sigma-Aldrich, Germany) was re-crystallized from ethanol before use. Bisphenol-A and epichlorohydrin were purchased from G. S. Chemical, India. Bisphenol-A was re-crystallized using toluene before use. Poly(amido-amine) (HY840, amine value 5–7 eq. kg−1) hardener for hyperbranched epoxy resin was obtained from Ciba Giegy, India. All other reagents used in the present investigation were of reagent grade.
2.2. Preparation of hyperbranched epoxy resin and its nanocomposites with carbon dot
Hyperbranched epoxy resin was synthesized by the polycondensation reaction of bisphenol-A and pentaerythritol (10 wt% with respect to bisphenol-A) with epichlorohydrin (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mole ratio with respect to hydroxyl group) at 110 °C with continuous stirring, as described in our recent report.27 An amount of 10.0 g (0.0438 mol) of bisphenol-A, 1.0 g (0.0073 mol) of pentaerythritol and 21.64 g (0.2339 mol) of epichlorohydrin were taken in two necked round bottom flask equipped with a water condenser and a dropping funnel. The reaction mixture was stirred with a magnetic stirrer. To this reaction mixture 5 N aqueous solution of NaOH (4.67 g, 0.1168 mol, equivalent to the hydroxyl group) was added slowly from a dropping funnel. After 4 h, the reaction was stopped and allowed to settle in a separating funnel to separate out the aqueous layer from the desired organic layer. Then the organic layer was washed with 15% aqueous sodium chloride solution followed by distilled water for 2–3 times. Then the epoxy resin was dried at 70 °C with the help of rotary evaporator (Eyela, Tokyo, Japan) to get viscous sticky transparent masses. This pristine hyperbranched epoxy (yield ∼ 97%) was coded as PHE and used as control for comparison purpose.
:2 mole ratio with respect to hydroxyl group) at 110 °C with continuous stirring, as described in our recent report.27 An amount of 10.0 g (0.0438 mol) of bisphenol-A, 1.0 g (0.0073 mol) of pentaerythritol and 21.64 g (0.2339 mol) of epichlorohydrin were taken in two necked round bottom flask equipped with a water condenser and a dropping funnel. The reaction mixture was stirred with a magnetic stirrer. To this reaction mixture 5 N aqueous solution of NaOH (4.67 g, 0.1168 mol, equivalent to the hydroxyl group) was added slowly from a dropping funnel. After 4 h, the reaction was stopped and allowed to settle in a separating funnel to separate out the aqueous layer from the desired organic layer. Then the organic layer was washed with 15% aqueous sodium chloride solution followed by distilled water for 2–3 times. Then the epoxy resin was dried at 70 °C with the help of rotary evaporator (Eyela, Tokyo, Japan) to get viscous sticky transparent masses. This pristine hyperbranched epoxy (yield ∼ 97%) was coded as PHE and used as control for comparison purpose.
The hyperbranched epoxy/carbon dot in situ nanocomposite was prepared by the same procedure using the same reactants and same amount under the same conditions. Here, only 1 wt% of carbon dot (with respect to the above synthesized resin) was incorporated with the 5 N aqueous solution of NaOH. The nanocomposite was coded as CHE.
The ex situ nanocomposite of hyperbranched epoxy with carbon dot (coded as ECP) was prepared by the solution technique with the help of mechanical shearing and ultrasonication similar to our earlier report.23 Ethanolic solution of carbon dot (1 wt% respect to hyperbranched epoxy) was added to the above synthesized hyperbranched epoxy resin (PHE) and the mixture was stirred mechanically for 4 h followed by ultrasonication (acoustic power density – 460 W cm−2) at 60% amplitude and 0.5 cycles for 10 min at room temperature.
2.3. Curing of the epoxy resin and the nanocomposites
PHE resin, CHE and ECP were cured by mixing homogenously with 50 phr of poly(amido-amine) hardener at room temperature in the presence of little amount of THF or ethanol solvent (0.5 mL g−1). Then the mixtures were uniformly coated on glass plates (75 × 25 × 1.3 mm3) and steel plates (150 × 50 × 1.6 mm3). The solvents and other volatiles were removed with the help of vacuum for 24 h and the plates were cured at 100 °C for 45 min. The optimization of curing reaction was done by determining swelling values (20–30%) of the cured films using THF after 48 h kept into glass bottles.
2.4. Characterization
The FTIR spectra were recorded on a Nicolet FTIR spectrometer (Impact-410, Madison, USA) using KBr pellet. The 1H NMR and 13C NMR spectra were recorded by a 400 MHz, Jeol FTNMR spectrometer using CDCl3 (1H NMR of PHE resin), d6-DMSO (PHE and CHE) and D2O (carbon dot) as the solvents and TMS as the internal standard. The morphologies and the selected area electron diffraction (SAED) patterns were analyzed by high resolution transmission electron microscope, HRTEM (JEOL, JEMCXII, Transmission Electron Microscope operating voltage at 200 kV). The physical properties such as epoxy equivalent, hydroxyl value and swelling value of the resins were measured by the standard test methods.19,20 The determination of shear viscosities of the resin and the nanocomposites were done by Bohlin rheometer CVO100 (Malvern, UK). The tensile strength (standard ASTM D 882) of the thermosets was measured by a Universal Testing Machine (UTM, WDW10, Jinan, China). The test was performed on rectangular film samples (size 60 × 10 × 0.3–0.4 mm3) with a 500 N load cell (crosshead speed of 10 mm min−1). Scratch hardness test (standard ASTM G171) was carried out by a scratch hardness tester (Sheen instrument Ltd, UK) on the surface of glass slide coated hyperbranched epoxy thermosets (area 75 × 25 × 0.3 mm3). Impact strength of the steel plate coated thermosets (area 150 × 50 × 1.6 mm3) was measured by an Impact tester (S. C. Dey Co., Kolkata) using the standard falling weight (ball) method (ASTM D 1709). The bending test of the thermosets (thickness of the films were 0.3–0.4 mm) was done using a mandrel with diameter 1 to 100 mm (standard ASTM D 522). All the tests for the measurement of mechanical properties were repeated for five times and average values were taken. Thermogravimetric analysis (TGA) of the thermosets was done using a PerkinElmer TG4000, US, with nitrogen flow rate of 30 mL min−1 and the heating rate of 10 °C min−1 from 30 to 700 °C. Differential scanning calorimetry (DSC) study of the thermosets was performed on a PerkinElmer DSC6000, US. The samples were heated at a rate of 10 °C min−1 and cooled at a rate of −10 °C under a nitrogen flow rate of 30 mL min−1 and a heating–cooling–heating cycle was followed. The UV-visible absorption spectra as well as percent of light transmittance by thermoset films (calculated from UV-visible spectra) were recorded using a UV spectrometer, Hitachi (U2001, Tokyo, Japan). The visual transparency of the thermoset films was checked by printed words covered with the thin thermosetting films (thickness 0.5 mm). The photoluminescent spectra of the resin and the nanocomposites (in ethanolic solution) as well as the thermoset films were recorded using a photoluminescent setup (PerkinElmer Singapore PTE Ltd, Singapore, Model LS 55). The optical color emission photos of the resin and nanocomposite; and the thermoset films were recorded by a UV fluorescence inspection cabinet (Labotech Solutions, Mumbai). The quantum yield values of carbon dot and in situ nanocomposite were determined at excitation wavelength of 360 nm by the following equation,| | |
QS = QR × IS/IR × AR/AS × ηS2/ηR2
| (1) |
where ‘Q’ is the quantum yield, ‘I’ is the intensity of luminescent spectra, ‘A’ is the absorbance at exited wavelength and ‘η’ is the refractive index of used solvent; using quinine sulfate (quantum yield 54) in 0.1 M H2SO4 solution as the reference. The subscripts ‘S’ for samples (carbon dot and nanocomposite) and ‘R’ for reference are used in this equation. The life time of the photoluminescence of carbon dot and nanocomposites in ethanol were analyzed by time resolved fluorescence using picosecond time-resolved cum steady state luminescence spectrometer (Edinburgh Instruments, UK, Model FSP920) at excitation wavelength 365 nm and emission wavelength 425 nm.
2.5. Cell proliferation and cytocompatibility assessment
Human dermal fibroblast (HDF) cells were cultured in high glucose Dulbecco's modified eagle medium (DMEM, Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA) antibiotics (Himedia, India), and 2 mM glutamine (Sigma-Aldrich, USA). Similarly, human keratinocyte, HaCaT cells were cultured in high glucose DMEM/F12 (Gibco, USA) supplemented with 10% FBS and antibiotics. For experiments, cells were maintained at 37 °C in humidified incubator with 5% CO2 and media was replenished twice a week. Cells were cultured on all the three variants of membrane namely, ex situ nanocomposite (ECP), in situ nanocomposite (CHE) and pristine epoxy thermoset (PHE). For cell proliferation studies, membranes with 10 mm diameter were pre-sterilized in laminar hood with 70% v/v ethanol for 4 h. Further, membranes were thoroughly washed with sterile phosphate-buffered saline (PBS, pH 7.4) to remove residual alcohol. Membranes were conditioned overnight in complete DMEM. HDF and HaCaT cells were seeded on sterilized membranes at a density of 5 × 104 cells per membrane. Cell proliferation/cytocompatibility was monitored with alamar blue dye reduction assay (Invitrogen, USA). Alamar blue is an aqueous fluorescent dye which measures the cell proliferation rate for the same batch of culture at different time points. It has no adverse effect on cellular phenotype, viability or proliferation. Alamar blue assay was done at specific intervals of 1, 4 and 7 days following manufacturers' protocol. Briefly, cell-seeded membranes were incubated in medium supplemented with 10% (v/v) alamar blue dye for 3 h. After incubation, 100 μL of solution from each sample was read at 570/600 nm in a multiplate reader (Tecan infinite M 200). Non-seeded membranes supplemented with 10% alamar blue dye were used as negative control.
2.6. Live/dead assay for cellular viability
Live/dead assay was performed to check the viability and adherence of cells on the membranes after 7 days of culture. Live and dead cells were stained by the staining solution containing 4 mM calcein AM and 2 mM ethidium homodimer (Sigma-Aldrich, USA), respectively. Briefly, cell-seeded membranes were rinsed with PBS (pH 7.4) and incubated in live/dead solution for 15–20 min in humidified incubator at 37 °C. Excess dye was washed with PBS and cells were visualized under florescent microscope (EVOS FL, Life Technologies, USA). Viable cells convert calcein AM to calcein which imparts green colour to the cells. Dead cells are visualized with ethidium homodimer which enters into dead cells and fluoresces red after binding with DNA.
Statistical analysis. All data are reported as mean ± standard deviation. For each experiment, n = 4 samples were used. Statistical analysis was performed by one-way analysis of variance (ANOVA). All statistical analyses were executed using OriginPro 8 and p < 0.05 was considered statistically significant and p < 0.01 as highly significant.
3. Results and discussion
3.1. Synthesis and characterization of the resin and nanocomposite
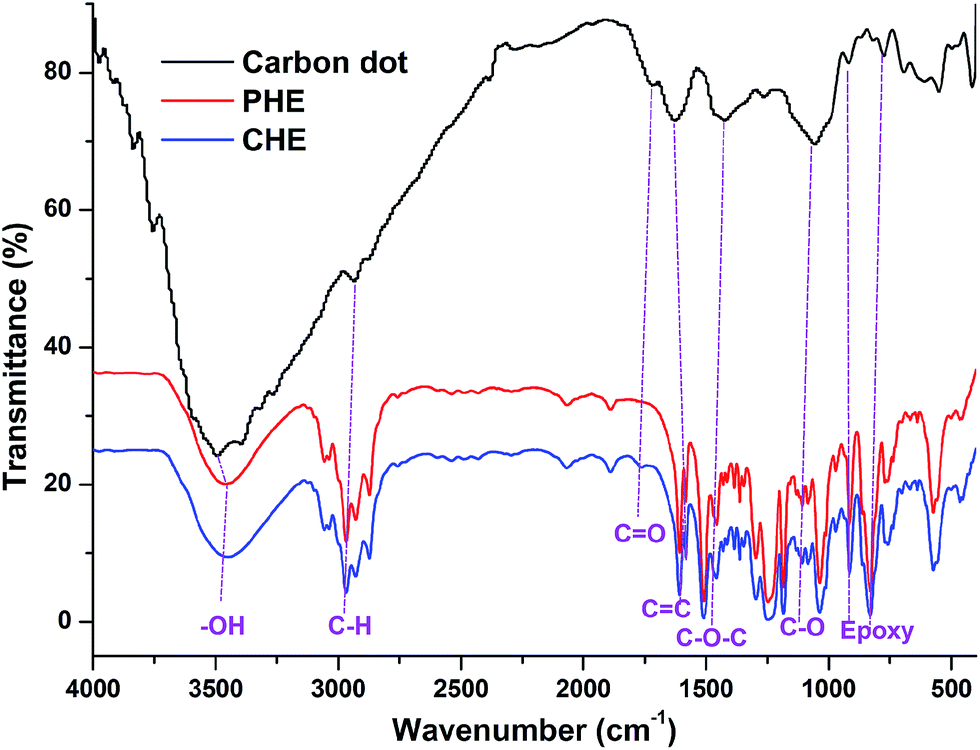
PHE resin was synthesized by an A2 + B4 polycondensation reaction. Initially, diglycidyl ether of bisphenol-A (A2) was formed in the reaction mixture as reactivity of bisphenol-A with epichlorohydrin is greater than pentaerythritol as the former contains aromatic proton. Finally, PHE resin was formed by reaction between diglycidyl ether of bisphenol-A (A2) and pentaerythritol (B4). On the other hand in case of CHE, carbon dot contains sodium salt of reactive phenolic and carboxylic acid groups as carbon dot was mixed with NaOH solution. The formation of sodium salt increases the reactivity of the phenolic and carboxylic groups by increasing nucleophilicity of the groups. Thus, here diglycidyl ether of bisphenol-A and epichlorohydrin were reacted with reactive carbon dot as well as pentaerythritol to form hyperbranched epoxy in situ nanocomposite as shown in Scheme 1. In this case the reaction of diglycidyl ether of bisphenol-A and epichlorohydrin with sodium salt of carbon dot followed the mechanism like anionic ring-opening polymerization.28 The structural features of PHE resin and CHE were characterized by FTIR and NMR studies which also confirmed that carbon dot participated in the reaction to form hyperbranched epoxy in situ nanocomposite. In the FTIR spectrum (Fig. 1) of PHE resin the stretching vibrations are attributed to the following features (νmax/cm−1): 919 and 835 (oxirane), 3457 (O–H), 3055 (aromatic C–H), 2970 (aliphatic C–H), 1606 (aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1450–1510 (C–O–C), 1244 (C–O) and 1035 (C–C).19,20 In case of carbon dot the stretching vibrations of FTIR spectrum are (νmax/cm−1): 3492 (–OH), 2935 (C–H), 1730 (CO), 1625 (aromatic CC), 1422 (C–O–C), 1264 (C–O) 918 and 780 (oxirane),23,26 whereas, in FTIR spectrum of CHE the stretching vibrations are attributed to the following features (νmax/cm−1): 919 and 835 (oxirane), 3444 (O–H), 3062 (aromatic C–H), 2971 (aliphatic C–H), 1764 (CO), 1460–1510 (C–O–C), 1614 (aromatic CC), 1244 (C–O) and 1037 (C–C). The hydroxyl and carboxylic acid stretching frequencies of carbon dot (at 3492 and 1730 cm−1 respectively) are shifted to 3444 and 1764 cm−1 in CHE due to their participation in different chemical reactions during formation of hyperbranched epoxy in situ nanocomposite as shown in Scheme 1a. In 1H NMR spectrum (Fig. 2a) of PHE resin, δH (400 MHz, CDCl3, Me4Si, ppm) values indicate the following structural features: 3.3 (1H, oxirane), 2.7 and 2.9 (2H, oxirane), 3.9 (2H, 4CH2 of pentaerythritol), 1.6 (3H, CH3 of bisphenol-A), 6.8 (4H, bisphenol-A), 7.1 (4H, bisphenol-A), 1.9 (1H, OH), 4.2 (1H, CHOH), 4.0 (2H, CH2 attached with oxirane), 3.9 (2H, CH2 attached with oxygen atoms of pentaerythritol unit) and 4.1 (2H, CH2 attached with oxygen atoms of bisphenol-A unit). Whereas, in 1H NMR spectrum (Fig. 2b) of CHE the same δH (400 MHz, d6-DMSO, Me4Si, ppm) values are present along with 5.7–6.2 (aromatic protons of carbon dot), 4.8–5.2 (HCCH of carbon dot), 3.6 (protons attached with ether of carbon dot) and 1.0–1.4 (sp3 protons of carbon dot) which are also present in 1H NMR spectrum of pristine carbon dot (Fig. 2c). The carboxylic acid and phenolic protons of carbon dot in the range of 7.5–9.5 ppm are not present in 1H NMR spectrum of CHE which confirms that these functional groups were participated in the reaction during preparation of CHE in situ nanocomposite. In 13C NMR spectrum (Fig. 3a) of PHE resin, the δC (d6-DMSO, ppm) values were observed at 43 (CH2, oxirane), 49 (CH, oxirane), 46–47 (central C of pentaerythritol unit), 114, 127, 143 and 156 (4C of bisphenol-A), 31 (CH3, bisphenol-A unit), 41 (C, isopropylidene of bisphenol-A unit), 68 (CH2, pentaerythritol unit) and 62–67 (CH2–O units and CHOH unit). All these δC (d6-DMSO, ppm) are present in 13C NMR spectrum (Fig. 3b) of CHE along with 20–30 (sp3 carbons of carbon dot), 65–68 (carbon dot carbons attached with ether linkages), 80–100 (sp2 carbons of carbon dot) and 170–180 (carbonyl carbons of carbon dot) which are also present in 13C NMR spectrum (Fig. 3c) of carbon dot. The degree of branching (DB) of PHE resin was determined from 13C NMR spectrum. A hyperbranched polymer contents three different types of units: dendritic (D), linear (L) and terminal (T) in its structure. DB is the ratio of the sum of integration of D and T units to the sum of integration of D, L, and T units, i.e., DB = (D + T)/(D + L + T).19,20 In case of PHE resin, the units with three and four, two and one hydroxyl group(s) of pentaerythritol substituted by diglycidyl ether of bisphenol-A moiety are representing dendritic (D), linear (L) and (T) units respectively. In 13C NMR spectrum the central carbon atom of pentaerythritol for these four units were observed at δ = 46.75, 46.55, 46.40 and 46.20 ppm respectively as shown in inset picture of Fig. 3a. The calculated DB from integration values of these peaks for PHE resin was found 0.78. However, in case of CHE structural characterization by DB is very difficult as carbon dot also took part in the reaction with in situ formed diglycidyl ether of bisphenol-A and other reactants as shown in Scheme 1. The calculated DB based on the above mentioned peaks was found to be 0.52, excluding the participation of carbon dot.
C), 1450–1510 (C–O–C), 1244 (C–O) and 1035 (C–C).19,20 In case of carbon dot the stretching vibrations of FTIR spectrum are (νmax/cm−1): 3492 (–OH), 2935 (C–H), 1730 (CO), 1625 (aromatic CC), 1422 (C–O–C), 1264 (C–O) 918 and 780 (oxirane),23,26 whereas, in FTIR spectrum of CHE the stretching vibrations are attributed to the following features (νmax/cm−1): 919 and 835 (oxirane), 3444 (O–H), 3062 (aromatic C–H), 2971 (aliphatic C–H), 1764 (CO), 1460–1510 (C–O–C), 1614 (aromatic CC), 1244 (C–O) and 1037 (C–C). The hydroxyl and carboxylic acid stretching frequencies of carbon dot (at 3492 and 1730 cm−1 respectively) are shifted to 3444 and 1764 cm−1 in CHE due to their participation in different chemical reactions during formation of hyperbranched epoxy in situ nanocomposite as shown in Scheme 1a. In 1H NMR spectrum (Fig. 2a) of PHE resin, δH (400 MHz, CDCl3, Me4Si, ppm) values indicate the following structural features: 3.3 (1H, oxirane), 2.7 and 2.9 (2H, oxirane), 3.9 (2H, 4CH2 of pentaerythritol), 1.6 (3H, CH3 of bisphenol-A), 6.8 (4H, bisphenol-A), 7.1 (4H, bisphenol-A), 1.9 (1H, OH), 4.2 (1H, CHOH), 4.0 (2H, CH2 attached with oxirane), 3.9 (2H, CH2 attached with oxygen atoms of pentaerythritol unit) and 4.1 (2H, CH2 attached with oxygen atoms of bisphenol-A unit). Whereas, in 1H NMR spectrum (Fig. 2b) of CHE the same δH (400 MHz, d6-DMSO, Me4Si, ppm) values are present along with 5.7–6.2 (aromatic protons of carbon dot), 4.8–5.2 (HCCH of carbon dot), 3.6 (protons attached with ether of carbon dot) and 1.0–1.4 (sp3 protons of carbon dot) which are also present in 1H NMR spectrum of pristine carbon dot (Fig. 2c). The carboxylic acid and phenolic protons of carbon dot in the range of 7.5–9.5 ppm are not present in 1H NMR spectrum of CHE which confirms that these functional groups were participated in the reaction during preparation of CHE in situ nanocomposite. In 13C NMR spectrum (Fig. 3a) of PHE resin, the δC (d6-DMSO, ppm) values were observed at 43 (CH2, oxirane), 49 (CH, oxirane), 46–47 (central C of pentaerythritol unit), 114, 127, 143 and 156 (4C of bisphenol-A), 31 (CH3, bisphenol-A unit), 41 (C, isopropylidene of bisphenol-A unit), 68 (CH2, pentaerythritol unit) and 62–67 (CH2–O units and CHOH unit). All these δC (d6-DMSO, ppm) are present in 13C NMR spectrum (Fig. 3b) of CHE along with 20–30 (sp3 carbons of carbon dot), 65–68 (carbon dot carbons attached with ether linkages), 80–100 (sp2 carbons of carbon dot) and 170–180 (carbonyl carbons of carbon dot) which are also present in 13C NMR spectrum (Fig. 3c) of carbon dot. The degree of branching (DB) of PHE resin was determined from 13C NMR spectrum. A hyperbranched polymer contents three different types of units: dendritic (D), linear (L) and terminal (T) in its structure. DB is the ratio of the sum of integration of D and T units to the sum of integration of D, L, and T units, i.e., DB = (D + T)/(D + L + T).19,20 In case of PHE resin, the units with three and four, two and one hydroxyl group(s) of pentaerythritol substituted by diglycidyl ether of bisphenol-A moiety are representing dendritic (D), linear (L) and (T) units respectively. In 13C NMR spectrum the central carbon atom of pentaerythritol for these four units were observed at δ = 46.75, 46.55, 46.40 and 46.20 ppm respectively as shown in inset picture of Fig. 3a. The calculated DB from integration values of these peaks for PHE resin was found 0.78. However, in case of CHE structural characterization by DB is very difficult as carbon dot also took part in the reaction with in situ formed diglycidyl ether of bisphenol-A and other reactants as shown in Scheme 1. The calculated DB based on the above mentioned peaks was found to be 0.52, excluding the participation of carbon dot.
 |
| | Scheme 1 (a) Chemical reactions of the reactants and carbon dot during the formation of CHE in situ nanocomposite and (b) its general structure. | |
 |
| | Fig. 1 FTIR spectra of carbon dot, PHE and CHE. | |
 |
| | Fig. 2 1H NMR spectra of (a) PHE, (b) CHE and (c) carbon dot. | |
 |
| | Fig. 3 13C NMR spectra of (a) PHE, (b) CHE and (c) carbon dot. | |
The epoxy equivalent, hydroxyl and viscosity values of PHE and CHE are given in Table 1. The numbers of epoxy as well as hydroxyl groups of the hyperbranched epoxy are higher for CHE than PHE due to participation of carbon dot in the former. As the number of these polar functional groups are higher in CHE compared to PHE resin, so the viscosity of the former is also higher (<1.5 folds), due to the presence of more intra- and inter-molecular attraction in CHE. Again, when carbon dot was physically mixed with PHE resin (i.e. in case of ECP) the viscosity increased more than four folds as the polar functional groups (hydroxyl, carboxylic acid, epoxy, ether, etc. as revealed from FTIR spectrum) of carbon dots are totally free to interact (by different physical interactions including polar–polar) with polar functional groups of PHE resin. The inner structure of CHE and ECP were examined by HR-TEM analysis. The uniform and homogenous dispersion of carbon dot inside CHE was observed in Fig. 4a. This uniform dispersion is due to the chemical bond formation of the functional groups of carbon dot during the preparation of hyperbranched epoxy. However in case of ECP, a few amounts of carbon dots are agglomerated inside the PHE matrix due to the intra molecular attraction of the polar functional groups of carbon dot as shown in Fig. 4b. Thus the interlayer spacing (d001) of carbon dot is more affected in case of CHE compared to ECP as shown in Fig. 4c–e. The graphitic d001 spacing of carbon dot increases from 0.24 to more than 1.0 nm after formation of CHE, whereas it increases up to 0.33 nm after formation of ECP. The poor crystalline structure of carbon dot becomes amorphous after formation of CHE as shown in selected area electron diffraction (SAED) patterns in Fig. 5. The bright spots in SEAD pattern of carbon dot (Fig. 5a) are completely disappeared from SAED pattern of CHE (Fig. 5c), whereas few bright spots are found in SAED pattern of ECP (Fig. 5b). This is due to the fact that after formation of CHE by the reaction of carbon dot with diglycidyl ether of bisphenol-A and pentaerythritol, the poor crystalline nature of carbon dot changes to completely amorphous state.
Table 1 The values of physical parameters for PHE, CHE and ECP
| Parameter |
PHE |
CHE |
ECP |
| Epoxy equivalent (g eq.−1) |
394 |
360 |
— |
| Hydroxyl value (mg KOH per g) |
100 |
144 |
— |
| Viscosity (Pa s) at 25 °C |
2.99 |
4.36 |
12.9 |
 |
| | Fig. 4 HR-TEM images of (a) CHE and (b) ECP at low magnification and internal structure of (c) carbon dot, (d) ECP and (e) CHE at very high magnification. | |
 |
| | Fig. 5 SAED patterns of (a) carbon dot, (b) ECP and (c) CHE. | |
3.2. Curing study
PHE resin, CHE and ECP were cured at 100 °C with 50 phr poly(amido-amine) hardener for 45 min and the swelling values of the thermosets are given in Table 2. PHE, CHE and ECP took 35, 30 and 45 min curing times respectively to reach <30% swelling value. However for better comparison all of them cured at same temperature (100 °C) for same time (45 min) with same amount of hardener (50 wt%) though the swelling values were kept in optimum (20–30%). The possible reactions occurred during the curing process were the reactions between amine groups of the hardener with the strained epoxy oxirane rings of PHE, CHE and ECP. Again, the hydroxyl, carboxylic acid and epoxy groups of carbon dot (in case of ECP) also took part in the curing reaction, however they took long time due to agglomeration of few amount of carbon dot inside PHE matrix. As CHE possessed highest number of epoxy groups (lowest epoxy equivalent, as given in Table 1), so it has the lowest swelling value. However, in case of ECP as carbon dot physically mixed with PHE, it exhibited the highest swelling value.
Table 2 Performance of PHE, CHE and ECP thermosets
| Parameter |
PHE |
CHE |
ECP |
| Calculated by integrating the area under stress–strain curves. Instrument limit of the scratch hardness was 10.0 kg (highest). Instrument limit of the impact strength was 100 cm (highest). Instrument limit of the mandrel diameter was 1 mm (lowest). |
| Swelling value (%) |
22 ± 0.2 |
21 ± 0.5 |
27 ± 0.8 |
| Tensile strength (MPa) |
51 ± 1.0 |
62.5 ± 2.0 |
44 ± 1.4 |
| Elongation at break (%) |
37 ± 0.5 |
45 ± 1.3 |
62 ± 2.5 |
| Toughnessa (MPa) |
1432 |
1682 |
2299 |
| Scratch hardnessb (kg) |
>10.0 |
>10.0 |
>10.0 |
| Impact resistancec (cm) |
>100 |
>100 |
>100 |
| Bending diameterd (mm) |
<1 |
<1 |
<1 |
| Initial degradation temperature (°C) |
279 |
291 |
235 |
3.3. Mechanical properties
Mechanical properties like tensile strength, elongation at break, toughness, scratch hardness, impact resistance and bending values of PHE, CHE and ECP thermosets are given in Table 2. The stress–strain profiles of the thermosets are shown in Fig. 6. From these results it is found that CHE thermoset possessed the highest tensile strength value. This is due to the fact that the aromatic carbonized carbon dot is chemically bonded (covalently bonded) with diglycidyl ether of bisphenol-A and pentaerythritol during the formation of hyperbranched epoxy in situ nanocomposite. This helps in strong interfacial interactions and uniform dispersion of carbon dot with compact structural architecture. It also exhibited higher elongation at break compared to PHE thermoset. This may be due to sliding of carbon dot layers at high stress as it is seen from Fig. 4e. Thus, the distance between the layers is increased after formation of CHE. This increased layer spacing and strong covalent bonds easily overcome the interlayer interactions between the carbon dot sheets. As CHE thermoset possessed higher tensile strength and elongation at break, it also showed higher toughness compared to PHE thermoset as calculated from the area under stress–strain curves (Fig. 6). However, tensile strength value of PHE thermoset is decreased after physically mixed with carbon dot in ECP thermoset. This is due to the fact that in case of ECP physical interactions of carbon with PHE are predominated over the chemical bond formation and carbon dots are agglomerated inside the PHE matrix as observed from the TEM image (Fig. 4b). Thus elongation at break and toughness values of PHE was increased after formation of ECP thermoset. PHE, CHE and ECP thermosets exhibited the same values of scratch hardness and impact resistance as the values reached the highest limit of the instruments for scratch hardness (10 kg) and impact resistance (100 cm). They also exhibited the lowest limit of the instrument for flexibility evaluation (1 mm bending diameter of mandrel) without any damage to the film.
 |
| | Fig. 6 Stress–strain profiles of PHE, CHE and ECP thermosets. | |
3.4. Thermal study
Initial degradation (5 wt% degradation) temperatures (°C) of the thermosets obtained from the TGA thermograms (Fig. 7), are given in Table 2. From the results, it was found that CHE thermoset exhibited the highest thermal stability as carbon dot chemically bonded with the structure of hyperbranched epoxy. Whereas, in case of physical mixture of carbon dot with PHE, the thermal stability of the thermoset was decreased as only physical interactions are present in ECP thermoset. However, maximum degradation of the thermosets were started after 300 °C and 50 wt% degradations of them were occurred at ∼390 °C as observed from TGA thermograms of the thermosets in Fig. 7. The thermosets were also degraded by two step patterns as observed in DTG curves (inset of Fig. 7), where the first step (∼360 °C) is due to the degradation of aliphatic moieties of hyperbranched epoxy and poly(amido-amine) hardener, and the second step (∼450 °C) is due to the degradation of aromatic moieties of hyperbranched epoxy and carbon dot. From the DSC study (Fig. 8), it was found that PHE, ECP and CHE thermosets exhibited close glass transition temperatures (Tg) due to their similar structural attributes. The slightly increment of Tg value of PHE after formation of carbon dot nanocomposite (CHE and ECP) is due to rigid structure of aromatic carbon dot as well as carbon dot restricted the chain motions of hyperbranched epoxy by different physico-chemical interactions. When, carbon dot act as an in situ reactive component in CHE, the effect is more due to covalent bond formation and thus, increment is higher compare to ex situ nanocomposite (ECP).
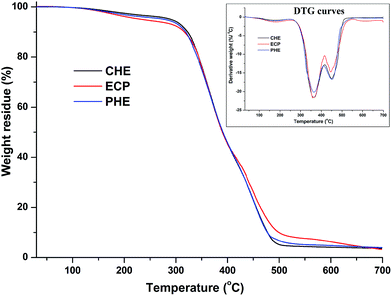 |
| | Fig. 7 TGA thermograms and DTG curves (inset figure) of CHE, ECP and PHE thermosets. | |
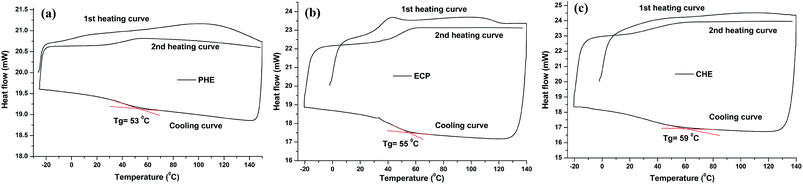 |
| | Fig. 8 DSC curves of (a) PHE, (b) ECP and (c) CHE thermosets. | |
3.5. Optical properties
The optical absorbance spectra of PHE, CHE and ECP thermosets are shown in Fig. 9a. The figure shows that the thermosets absorbed more amount of light in the UV region. In case of PHE thermoset, absorption of UV light is due to the π–π* transition of aromatic bisphenol-A moiety present in its structure. Whereas, in case of CHE and ECP thermosets absorption is due to the π–π* and n–π* transitions of carbon dot as well as the π–π* transition of aromatic bisphenol-A moiety.23,26 From the Fig. 9a, it is found that ECP thermoset absorbed the highest amount of UV light because of the presence of only physical interactions between carbon dot and PHE, and thus, carbon dots are free to interact with UV light. However, in case of CHE depletion of the electronic transition was occurred due to the participation of the functional groups of carbon dot in chemical reactions to form hyperbranched epoxy and thus, it absorbed less amount of UV light.
 |
| | Fig. 9 (a) Optical absorbance spectra and; (b) percent of transmittance and transparency (inset pictures) of PHE, ECP and CHE thermosets. | |
The visual transparency and percent of transmittance of visible light of PHE, CHE and ECP thermoset films are shown in Fig. 9b. In the inset picture it is found that CHE thermoset film showed the highest transparency. This is due to the quantum size carbon particles with large number of polar functional groups participate in the reaction to form the hyperbranched epoxy. Thus, the percent of transmittance of visible light was also the highest in case of CHE. However, the transparency as well as the percent of transmittance of visible light of PHE was decreased after formation of ECP. This is due to the presence of only physical interactions between carbon dot and PHE as well as agglomeration of carbon dot in the PHE matrix.
The photoluminescence (PL) behaviors of CHE and thermosetting solid film of CHE are shown in Fig. 10. CHE exhibited higher PL intensity as well as spectrum is also narrower compared to carbon dot at the same concentration in ethanolic solution (0.001%) as shown in Fig. 10a. This is due to uniform dispersion of carbon dot without agglomeration after formation of CHE. However, after formation of ECP these were not happened due to the presence of agglomerated carbon dot inside the PHE matrix. From the Fig. 10b, it was found that the intensity of the PL spectra increases with the increase of concentration of CHE in ethanolic solution. On the other hand, reverse trend was found in case of carbon dot in our earlier study,26 because of agglomeration of free carbon dot at higher concentration due to interactions among the polar functional groups on the surface. Whereas, these groups are chemically bonded with other reactants during the formation of CHE and thus, in this case homogeneous and uniform dispersion of carbon dot was observed without any agglomeration. However, CHE also exhibited excitation wavelength dependent PL behavior like carbon dot as shown in Fig. 10c. Here, also the highest intensity PL spectrum was found at 360 nm excitation wavelength similar to carbon dot as reported in our earlier study.26 This is common PL behavior of carbon dot and the reasons for this behavior are the presence of different particle sizes and the distribution of the different surface energy traps of the carbon dot as reports earlier as well as in literatures.26,29–31 Quantum yield of CHE was found 12.69 using quinine sulfate as the reference according to eqn (1) on excitation at 360 nm wavelength, which was higher compared to carbon dot (8.95) as measured in our earlier study.26 This is because of the uniform and homogenous dispersion of carbon dot inside the matrix in CHE compared to bare carbon dot. Second order exponential fitting curves of fluorescence decay of carbon dot, CHE and ECP are shown in Fig. 11. Here, the fitting curves and the life time of PL values are comparable with the reported carbon dot as well as carbon dot based nanocomposites.32–34 The PL spectra of CHE, ECP and PHE thermoset films are shown in Fig. 10d. From the spectra, it was found that CHE exhibited the highest PL intensity due to strong interfacial interaction of carbon dot with hyperbranched polymer as well as homogeneous and uniform dispersion of carbon dot inside the matrix as stated above. However, PL intensity of PHE was not significantly improved after formation of ECP as only physical interactions are present and agglomeration of carbon dot was also formed inside the PHE matrix. PHE thermoset possessed negligible PL due to the presence of aromatic bisphenol-A moiety in its structure.
 |
| | Fig. 10 PL spectra of (a) CHE, ECP and carbon dot in ethanolic solution at same concentration (0.001%); (b) CHE (in ethanolic solution) with variation of concentration and (c) variation of excitation wavelength; and (d) CHE, ECP and PHE thermosets. | |
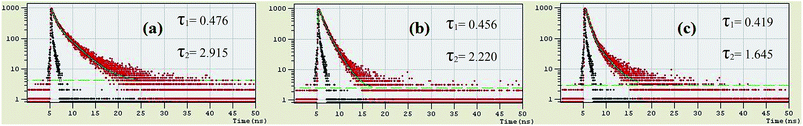 |
| | Fig. 11 Fluorescence decay profiles of (a) carbon dot, (b) CHE and (c) ECP. | |
The optical color emissions of carbon dot, CHE as well as CHE and ECP thermosets at different wavelengths of UV light are shown in Fig. 12. From Fig. 12a–c it was found that CHE as well as CHE thermoset emits green and light blue fluorescence similar to carbon dot after illumination with short (254 nm) and long (365 nm) wavelengths of UV light. The green and blue color emissions are due to the corresponding band gaps of carbon dot at different excitation wavelengths of UV light and the color change is due to the presence of different sizes of carbon dot as reported earlier.23 ECP thermoset also emits similar color like CHE thermoset (Fig. 12c), however due to some agglomeration of carbon dot inside the PHE matrix brightness of color is inferior to CHE thermoset.
 |
| | Fig. 12 Optical color emissions of (a) carbon dot, (b) CHE and; (c) CHE and ECP thermosets under exposure of UV lights at 254 and 365 nm. | |
3.6. Cell proliferation and cytocompatibility assessment
In vitro cell proliferation/cytocompatibility studies were performed on ECP, CHE and PHE membranes using HDF and HaCaT cells. Alamar blue assay was done to quantify cell viability and proliferation over time to assess cytocompatibility of the membranes. Increased reduction percentage of alamar blue overtime relates to higher number of proliferating cells which in turn indicates cytocompatibility of the membranes favoring cell growth.35–37 All the membranes showed cell proliferation and increase in cell number with time. After 7 days of culture, HDF cell proliferation showed the following trend: ∼82% on CHE, ∼44% on ECP and ∼16% on PHE compared to day 1 of culture (Fig. 13a) (p < 0.01). On day 7, all the membranes CHE, ECP and PHE exhibited significant differences for HDF cells (p < 0.01). In case of HaCaT cells, similar proliferation trend was observed (Fig. 13b). On day 7, HaCaT cells proliferated ∼3.17 fold on CHE, ∼2.47 fold on ECP and ∼2.55 fold on PHE as compared to culture day 1. For HaCaT cells there was a significant difference between CHE with ECP and CHE with PHE, respectively (p < 0.01). However, no significant difference was observed between ECP and PHE (p > 0.05). Thus CHE is the best material for HDF and HaCaT cell proliferation may be due to better compatibility compared to other two materials.
 |
| | Fig. 13 Alamar blue cell proliferation assay showing (a) human dermal fibroblast (HDF) and (b) skin keratinocyte (HaCaT) cell proliferation on all the three membranes. Data represented as average ± standard deviation (**p < 0.01). | |
3.7. Live/dead assay for cellular viability
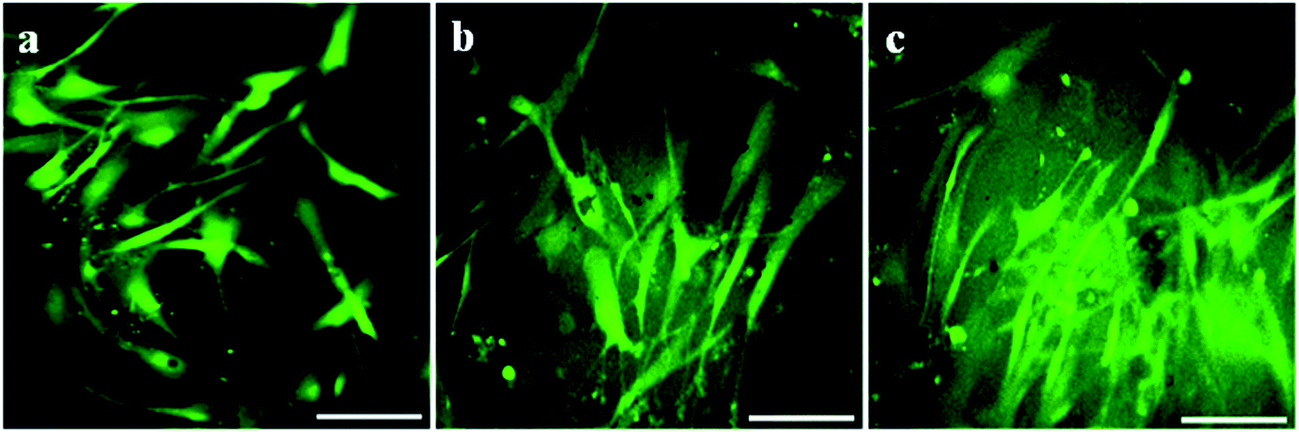
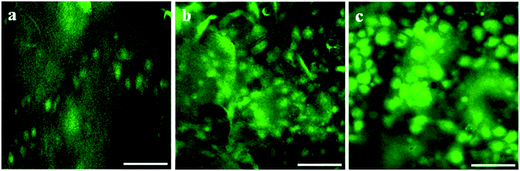
As evident from fluorescent microscopic images both HDF and HaCaT cells attached firmly to all the membranes and proliferated well (Fig. 14 and 15). Cells maintained native spindle morphology suggesting the membranes to be cytocompatible and favoring growth and attachment. Cells were evenly distributed on the membranes. Hence, the viability, spreading and proliferation of skin fibroblasts and keratinocytes on these membranes suggest them to be highly applicable as a bio-sealant material in surgical wound skin closure.
 |
| | Fig. 14 Fluorescent microscopic images showing human dermal fibroblast (HDF) cells growing on membranes after seven days of culture. (a) Ex situ (ECP), (b) in situ (CHE) and (c) pristine (PHE). Scale bar represents 100 μm. | |
 |
| | Fig. 15 Fluorescent microscopic images showing human skin keratinocyte (HaCaT) cells growing on membranes after seven days of culture. (a) Ex situ (ECP), (b) in situ (CHE) and (c) pristine (PHE). Scale bar represents 100 μm. | |
Furthermore, this biocompatibility is helpful for safe handling of the material for its different applications. The nanocomposite exhibited good biocompatibility with human skin fibroblasts and keratinocytes cells as observed in Fig. 13–15 with good viability, spreading and proliferation of these cells on it. The thermoset contains 50 wt% vegetable oil derived poly(amido-amine) hardener, which is an excellent biodegradable material as shown in our earlier works.20,38–40 A very recent study of our group also showed that an epoxy thermoset with similar structural attributes and same hardener was degraded under physiological conditions and the degraded products were nontoxic.40 Thus, it is expected that the studied material may be safe to use and partially biodegradable.
4. Conclusion
The present study, therefore, demonstrated an excitation wavelength dependent photoluminescence biocompatible carbon dot/hyperbranched epoxy in situ nanocomposite. NMR and TEM studies confirmed the participation of carbon dot in chemical reaction during the formation of hyperbranched epoxy in situ nanocomposite. The study also revealed superior mechanical, thermal, optical and bio-sealant properties of this nanocomposite over the ex situ nanocomposite of carbon dot with hyperbranched epoxy. These results clearly evident the incorporation of carbon dot in the hyperbranched structure through covalent linkages. Thus, the study may open up a new avenue for high performance transparent fluorescent polymeric materials used in advanced optical as well as bio-sealant applications.
Acknowledgements
Dr B. B. Mandal greatly acknowledges to Government of India for financial support by the following grants: DBT-BT/505/NE/TBP/2013 and DBT-BT/IN/Sweden/38/BBM/2013 for biological works of the present study.
References
- S. V. Gaponenko, Optical Properties of Semiconductor Nanocrystals, Cambridge University Press, Cambridge, 1998 Search PubMed.
- N. Tomczak, D. Janczewski, M. Han and G. J. Vancso, Prog. Polym. Sci., 2009, 34, 393–430 CrossRef CAS PubMed.
- X. Li, Y. Wu, D. Steel, D. Gammon, T. H. Stievater, D. S. Katzer, D. Park, C. Piermarocchi and L. J. Sham, Science, 2003, 301, 809–811 CrossRef CAS PubMed.
- J. H. Park, J. Y. Kim, B. D. Chin, Y. C. Kim, J. K. Kim and O. O. Park, Nanotechnology, 2004, 15, 1217–1220 CrossRef CAS.
- N. Tomczak, R. Liua and J. G. Vancso, Nanoscale, 2013, 5, 12018–12032 RSC.
- M. D. Goodman, J. Xu, J. Wang and Z. Lin, Chem. Mater., 2009, 21, 934–938 CrossRef CAS.
- H. Althues, P. Simon and S. Kaskel, J. Mater. Chem., 2007, 17, 758–765 RSC.
- L. Chen, C. Wang, Q. Li, S. Yang, L. Hou and S. Chen, J. Mater. Sci., 2009, 44, 3413–3419 CrossRef CAS.
- Q. Li, T. Y. Ohulchanskyy, R. Liu, K. Koynov, D. Wu, A. Best, R. Kumar, A. Bonoiu and P. N. Prasad, J. Phys. Chem. C, 2010, 114, 12062–12068 CAS.
- C. S. Stan, C. Albu, A. Coroaba, M. Popa and D. Sutiman, J. Mater. Chem. C, 2015, 3, 789–795 RSC.
- S. T. Yang, L. Cao, P. G. Luo, F. Lu, X. Wang, H. Wang, M. J. Meziani, Y. Liu, G. Qi and Y. P. Sun, J. Am. Chem. Soc., 2009, 131, 11308–11309 CrossRef CAS PubMed.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS PubMed.
- Z. A. Qiao, Y. Wang, Y. Gao, H. Li, T. Dai, Y. Liu and Q. Huo, Chem. Commun., 2010, 46, 8812–8814 RSC.
- L. Zhou, B. He and J. Huang, Chem. Commun., 2013, 49, 8078–8080 RSC.
- Y. Hao, Z. Gan, J. Xu, X. Wu and P. K. Chu, Appl. Surf. Sci., 2014, 311, 490–497 CrossRef CAS PubMed.
- W. Kwon, S. Do, J. Lee, S. Hwang, J. K. Kim and S. W. Rhee, Chem. Mater., 2013, 25, 1893–1899 CrossRef CAS.
- T. Li, J. Zhang, H. Wang, Z. Hu and Y. Yu, ACS Appl. Mater. Interfaces, 2013, 5, 8968–8981 CAS.
- J. C. Huang, Y. P. Chu, M. Wei and R. D. Deanin, Adv. Polym. Technol., 2004, 23, 298–306 CrossRef CAS PubMed.
- B. De and N. Karak, J. Mater. Chem. A, 2013, 1, 348–353 CAS.
- B. De, K. Gupta, M. Mandal and N. Karak, ACS Sustainable Chem. Eng., 2014, 2, 445–453 CrossRef CAS.
- W. Zou, Z. Du, H. Li and C. Zhang, J. Mater. Chem., 2011, 21, 13276–13282 RSC.
- Y. Yang, Y. Q. Li, S. Y. Fu and H. M. Xiao, J. Phys. Chem. C, 2008, 112, 10553–10558 CAS.
- B. De, B. Voit and N. Karak, ACS Appl. Mater. Interfaces, 2013, 5, 10027–10034 CAS.
- P. Zhang, W. Li, X. Zhai, C. Liu, L. Dai and W. Liu, Chem. Commun., 2012, 48, 10431–10433 RSC.
- D. Zhang, E. Liang, T. Li, S. Chen, J. Zhang, X. Cheng, J. Zhou and A. Zhang, RSC Adv., 2013, 3, 9522–9529 RSC.
- B. De and N. Karak, RSC Adv., 2013, 3, 8286–8290 RSC.
- B. De and N. Karak, RSC Adv., 2015, 5, 35080–35088 RSC.
- A. Goodwin and D. Baskaran, Macromolecules, 2012, 45, 9657–9665 CrossRef CAS.
- S. Sahu, B. Behera, T. K. Maiti and S. Mohapatra, Chem. Commun., 2012, 48, 8835–8837 RSC.
- H. Li, Z. Kang, Y. Liu and S. T. Lee, J. Mater. Chem., 2012, 22, 24230–24253 RSC.
- Y. Wang and A. Hu, J. Mater. Chem. C, 2014, 2, 6921–6939 RSC.
- D. Dey, T. Bhattacharya, B. Majumdar, S. Mandani, B. Sharma and T. K. Sarma, Dalton Trans., 2013, 42, 13821–13825 RSC.
- H. Choi, S. J. Ko, Y. Choi, P. Joo, T. Kim, B. R. Lee, J. W. Jung, H. J. Choi, M. Cha, J. R. Jeong, I. W. Hwang, M. H. Song, B. S. Kim and J. Y. Kim, Nat. Photonics, 2013, 7, 732–738 CrossRef CAS PubMed.
- J. Gu, D. Hu, W. Wang, Q. Zhang, Z. Meng, X. Jia and K. Xi, Biosens. Bioelectron., 2015, 68, 27–33 CrossRef CAS PubMed.
- B. B. Mandal, A. Grinberg, E. S. Gil, B. Panilaitis and D. L. Kaplan, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7699–7704 CrossRef CAS PubMed.
- B. B. Mandal and S. C. Kundu, Biomaterials, 2009, 30, 2956–2965 CrossRef CAS PubMed.
- B. B. Mandal and S. C. Kundu, Acta Biomater., 2009, 5, 2579–2590 CrossRef CAS PubMed.
- B. Roy, P. Bharali, B. K. Konwar and N. Karak, Int. J. Mater. Res., 2014, 105, 296–307 CrossRef CAS.
- R. Duarah and N. Karak, RSC Adv., 2015, 5, 64456–64465 RSC.
- S. Barua, P. Chattopadhyay and N. Karak, J. Mater. Chem. B, 2015, 3, 5877–5885 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.