
Adsorption of quinoline from liquid hydrocarbons on graphite oxide and activated carbons†
Xiao Fenga,
Xiaoliang Mab,
Na Li*a,
Chao Shanga,
Xiaoming Yanga and
Xiao Dong Chen*a
aSchool of Chemical and Environmental Engineering, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, Jiangsu 215123, China. E-mail: chemlina@suda.edu.cn; xdchen@suda.edu.cn
bPetroleum Research Center, Kuwait Institute for Scientific Research, Safat 13109, Kuwait
First published on 24th August 2015
Abstract
Four carbon-based adsorbents (activated carbon, oxidatively modified activated carbon, graphite, and graphite oxide) were investigated as adsorbents for selectively removing quinoline from a model hydrocarbon fuel. The surface chemical properties of these carbon-based adsorbents were characterized by temperature-programmed desorption coupled with mass spectrometry (TPD-MS), X-ray photoelectron spectroscopy (XRD), elementary analysis (EA) and nitrogen adsorption–desorption analysis in detail. The influences of the textural structures and the surface functional groups of these carbon adsorbents on their adsorption performance were examined. The activated carbon modified by ammonium persulfate oxidation (APS) can achieve an adsorption capacity as high as 35.7 mg-N g−1. The results indicated that the oxygen-containing functional groups on the surface play a crucial role in determining their adsorptive performance for quinoline. In addition, enhancement of the interlayer distance in the graphite oxide results in a dramatic increase in the adsorption capacity of the graphite oxide. The accessibility of the oxygen functional groups on the surface for quinoline is important to the adsorption behavior. Considering its high adsorption capacity and good regenerability, the graphite oxide may also be a promising adsorbent for selectively adsorptive removal of nitrogen compounds from liquid hydrocarbon streams.
1. Introduction
In-depth denitrogenation and desulfurization of liquid hydrocarbon streams for producing ultra-clean fuels have attracted considerable attention worldwide due to the stringent requirement on the basis of environmental protection regulations. The sulfur and nitrogen compounds in fuels are converted to SOx and NOx, respectively, during combustion, which are responsible for global warming, acid rain, fog and haze.1–3 Thus, the fuels with ultra-low sulfur and ultra-low nitrogen compounds are required worldwide.4,5 Hydrodesulfurization (HDS) is the most important process for producing ultra-low sulfur transportation fuels. The presence of nitrogen compounds in the fuels, even at low concentrations, could strongly inhibit the HDS process.6–8 The removal of nitrogen compounds before HDS could significantly improve the HDS performance and lower the sulfur level to less than 5 ppm.6,9,10 Such a process is especially required for the feedstocks that contain high nitrogen compounds, such as the coal-derived liquid. The conventional method of removing nitrogen compounds is via hydrodenitrogenation (HDN) in oil refineries, which is more difficult than HDS, the HDN process requires severe reaction conditions, such as elevated temperature and pressure with higher hydrogen consumption.11–13 Therefore, some non-hydroprocessing approaches, such as adsorptive denitrogenation (ADN), oxidative denitrogenation (ODN), and extractive denitrogenation (EDN) have been proposed.6,9,14–16Among them, ADN has attracted considerable attention because it can be performed under ambient conditions without using hydrogen.6,14–17 An efficient adsorbent plays a crucial role in the adsorption approach. A good adsorbent for this application should have a sufficiently high capacity and high selectivity towards nitrogen compounds with good stability in the process, as well as easy preparation, modification and regeneration. Therefore, a wide range of adsorbents have been developed and tested for ADN, such as metal oxides, activated carbon, ion-exchange resins, and supported metal (Ni/SiO2–Al2O3), activated alumina, zeolites, metal organic frameworks (MOFs) and others.6,14–16,18
Among the adsorbents, activated carbon (AC) has been widely studied in ADN due to its significant advantages, such as lower price, wider range of sources, and easy modification of pore structure and surface functional groups.6,15,16,18 More importantly, it has good capacity and reasonable selectivity for nitrogen compounds. However, the maximum ADN capacity of AC reported in the literature is only around 17.6 mg-N per gram of solid,15,19 and the adsorption selectivity of ACs for the nitrogen compounds, especially those in real fuels remains limited.20,21 The adsorption behavior of carbon materials depends on both the chemical properties, such as the oxygen-containing functional groups (OCFGs) on their, and the physical properties such as the pore structure and surface area. The mechanisms proposed by different researchers vary considerably.16,22,23 According to the previous studies, the relationship between the properties of AC and the adsorption capacity for both quinoline and indole has not clearly established yet.14,15 The chemical properties of the ACs, such as the concentration of OCFGs, shown in Fig. 1a, have been expected to play important roles in determining the adsorption capacity and selectivity for nitrogen compounds, especially for quinoline.24 The commercially available ACs and their modified versions used in the previous studies showed different chemical properties and physical structures, which make it difficult to discern their individual effects on their adsorption performances.9,23,25
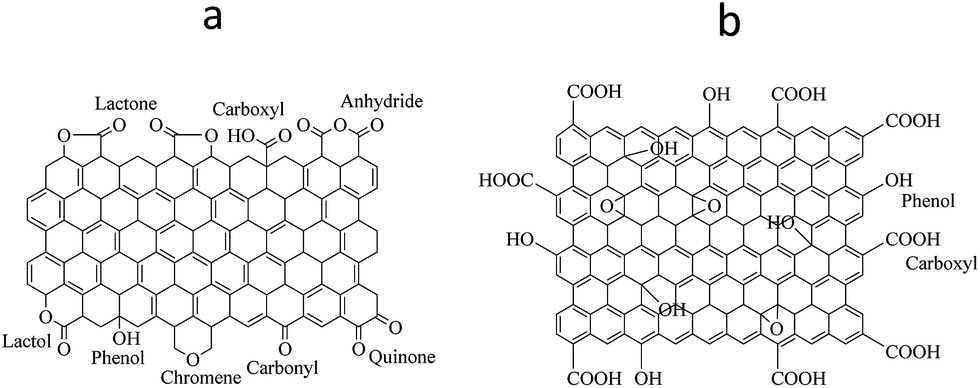 | ||
| Fig. 1 Oxygen groups on (a) activated carbon and (b) graphite oxide. | ||
Alternatively, graphite oxide (GO), as an emerging class of carbon materials, has regained the great attention due to its role as a precursor for the cost-effective mass production of graphene.26–29 GO can be prepared by the severe oxidation of graphite (GF). The oxidation process can introduce a large amounts of oxygen groups in the forms of carboxyl groups, hydroxyl, epoxides, and ketones groups, as shown in Fig. 1b.27,30,31 In previous studies, GO samples with low surface areas (e.g., below 20 m2 g−1) and non-porous structures were prepared.32 The samples had higher concentrations of the oxygen groups than the oxidative activated carbon (OAC), which may make the GO more suitable for conducting fundamental research to obtain insights into the ADN mechanisms, although it is a disadvantage for many applications. This property allows ones to focus on examining the influence of the OCFGs without or with less disturbance from the textural structures.
Recently, GO has been successfully used as an adsorbent in both gas and liquid-phase separation.33–35 Ren et al. reported that GO had an significant adsorption capacity for Cu(II) due to its high concentration of surface acidic oxygen groups, and demonstrated the usage of GO for adsorption removal of methylene blue dye. This report indicates that the GO might be a potential adsorbent for adsorptive removal of some polar compounds, such as nitrogen-containing compounds, from liquid hydrocarbon streams.36 To the best of our knowledge, the study of ADN on GOs has not been reported in the available literature yet.
In this work, the adsorption behavior of GF, GO, AC and OAC with different physical and chemical properties were studied. Quinoline, which is one of the main basic nitrogen compounds in fossil fuels, especially in coal liquid, was used as an adsorbate in this study.37 The adsorption capacities of different carbon adsorbents for quinoline in a model fuel were measured to fundamentally understand the effects of the textural structures and OCFGs of different adsorbents on their ADN performances, and to clarify their similarities and differences in application for the adsorptive removal of quinoline from liquid hydrocarbons.
2. Experimental section
2.1. Carbon adsorbents and oxidation treatment
A commercial AC (TF-B520) was provided by the Shanghai Sino Tech Investment Management Company. To introduce the OCFGs on the surface, AC was chemically modified by treating it with 1.5 mol L−1 ammonium persulfate (APS) solution (in 1 mol L−1 H2SO4). About 1.0 g of AC was added to 20 mL of APS solution in a beaker held at 60 °C and for 3 h, as described in detail previously.24 The modified sample was collected by filtration and washed with abundant deionized water till reaching neutral. The treated AC was dried under vacuum at 120 °C overnight before use. This oxidized AC is denoted as OAC. Nearly no functional groups other than oxygen-containing groups and metal ions can be introduced onto the AC surfaces according to our previous work.15GO was prepared from commercial GF (Alfa Aesar, natural 325 mesh) using the Hummers method.38 GF (3.0 g) and NaNO3 (3.0 g) were dispersed in 69 mL of 98% H2SO4 in a beaker held in an ice bath. KMnO4 (9.0 g) was slowly added into this mixture. After stirring for 10 min in the ice bath, the beaker was transferred to a 35 °C water bath for 1 h, and then the deionized water (138 mL) was slowly added to the suspension. After increasing the temperature to 90 °C and holding it for 15 min, the suspension was diluted with 420 mL of deionized water, and then 30% H2O2 (5 mL) was added. The mixture was kept at room temperature overnight. The formed GO that had deposited at the bottom was separated from the excess liquid by decantation. The concentrated solution was dialyzed with deionized water for 72 h to remove the metal ions. The concentrated solution was further diluted to 2.0 L and then treated ultra-sonically for 1 h before being spray dried to obtain the GO.
2.2. Characterization of textural properties of the adsorbents
The textural properties of the carbon materials were determined via the nitrogen adsorption/desorption technique at 77 K using the ASAP 2020 surface area and porosimetry analyzer. The standard BET and DFT models were applied to determine the surface area and pore volume.The characteristics of the structure were also conducted by using X-ray diffraction technique (XRD). The powdered samples were kept in a 2.5 cm × 2.5 cm × 1 mm quartz block. The powder was pressed onto the quartz block using a glass slide to obtain a uniform distribution. The data were obtained using a X'Pert-Pro MPD (PANalytical B.V.) and a Cu Kα radiation source with a scan range of 5–70° at a rate of 1° min−1.
2.3. Surface OCFGs of the carbon adsorbents
The types and concentrations of the OCFGs on the surfaces of the carbon adsorbents were determined using the temperature-programmed desorption method (Chem BET plus Quantachrome) with a mass spectrometer detector (Dycor, Model 2000, TPD-MS). These surface OCFGs were identified and quantified using the modified TPD-MS deconvolution method, which was developed in a previous study through modification of the method reported by Figueiredo et al.392.4. Batch adsorption
The liquid phase adsorption was performed for 3 h in 40 mL plastic vials filled with 0.02 g of adsorbent and desired weight of the model fuel (MF) which contains quinoline in heptane with a concentration of 300 ppmw as nitrogen. After the desired time was reached, the mixture was separated by centrifuging at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min. The treated MF was analyzed to estimate the adsorption capacity. The Langmuir isotherm equations have been widely used to analyze equilibrium adsorption data. The linear form of the Langmuir is shown as follows:
000 rpm for 15 min. The treated MF was analyzed to estimate the adsorption capacity. The Langmuir isotherm equations have been widely used to analyze equilibrium adsorption data. The linear form of the Langmuir is shown as follows:| ce/qe = 1/Kqm + ce/qm | (1) |
The procedure of kinetic tests was basically identical to that of equilibrium tests.40,41 The experiment was carried out with an adsorbent-to-MF weight ratio of 1:500 at 25 °C. The samples were taken at regular time intervals. The concentration of nitrogen in the treated MF was measured.
2.5. Analysis of the treated model fuel
The concentration of quinoline in the treated MF was quantitatively analyzed using a GC 2010 (Shimadzu) plus gas chromatography with a low-polarity capillary column (Rtx®-5, 30 m × 0.32 mm × 0.25 μm, RESTEK) and a nitrogen/phosphorous detector (NPD). The oven temperature was initially set at 130 °C, ramped at 20 °C min −1 to 280 °C, and then held at this temperature for 5 min. The temperatures of injector and detector were at 280 °C and 300 °C, respectively.2.6. Regeneration of adsorbent
Spent GO adsorbents were regenerated by ultrasound treatment for 1 h in different solvents, including acetone, ethanol, toluene, and N,N-dimethylformamide (DMF).42,43 The regenerated GO adsorbents were dried at 60 °C in vacuum before the following adsorption test.3. Results and discussion
3.1. Textual properties of the adsorbents
The textual properties of four carbon adsorbents, AC, OAC, GF and GO, were characterized by the nitrogen adsorption–desorption technique. The nitrogen adsorption–desorption isotherms are shown in Fig. 2. The OAC shows a significant reduction in N2 uptake compared with the original AC. The BET surface area of the OAC was deceased by 50% after 3 h of oxidation due to the partial destruction or collapse of the porous structure of the carbon during the oxidation process.15,24 | ||
| Fig. 2 Nitrogen adsorption–desorption isotherms of AC, OAC, GF, and GO. | ||
GO exhibits a slightly higher nitrogen adsorption capacity (9.4 m2 g−1) than that of GF (5.9 m2 g−1), although it is much lower than that of AC and OAC. Its lower surface area might be ascribed to the agglomeration of GO layers during drying process. The significant amount of oxygen groups resulted in the agglomeration due to the strong H-bonding between GO layers.27,30 The N2 molecules might be difficult to insert into the interlayer space of GO, but can only adsorb on the external surface. Further investigations are necessary to clarify it. The pore size distributions of all of the adsorbents are shown in Fig. S1.† A significant reduction of the pore volume was observed for AC after the oxidation modification, e.g. from 0.99 cm3 g−1 to 0.46 cm3 g−1. The detailed physical properties of AC, OAC, GF and GO are summarized in Table 1.
| Sample | SBET (m2 g−1) | Vtotal (cm3 g−1) |
|---|---|---|
| AC | 2051.5 | 0.99 |
| OAC | 1028.9 | 0.46 |
| GF | 5.9 | — |
| GO | 9.4 | — |
3.2. Characterization of the OCFGs on adsorbent surface
The chemical properties of AC, OAC, GF and GO, e.g. total oxygen concentrations and distribution of OCFGs on the surface, were determined by various techniques. The TPD-MS was used to monitor the CO2 and CO released during the temperature program to obtain a deeper insight into the surface chemistry of the adsorbents. Fig. 3 shows the CO, CO2 and H2O TPD profiles for OAC and GO. Significant amounts of CO and CO2 were released when the temperature was higher than 200 °C, due to the decomposition of OCFGs on OAC and GO. The amounts of CO and CO2 released and the total oxygen contents for the different samples were calculated using the methods reported in the literatures.24,39 The physical adsorbed water and the water formed during the decomposition of oxygen groups were also calculated. The results are summarized in Table 2.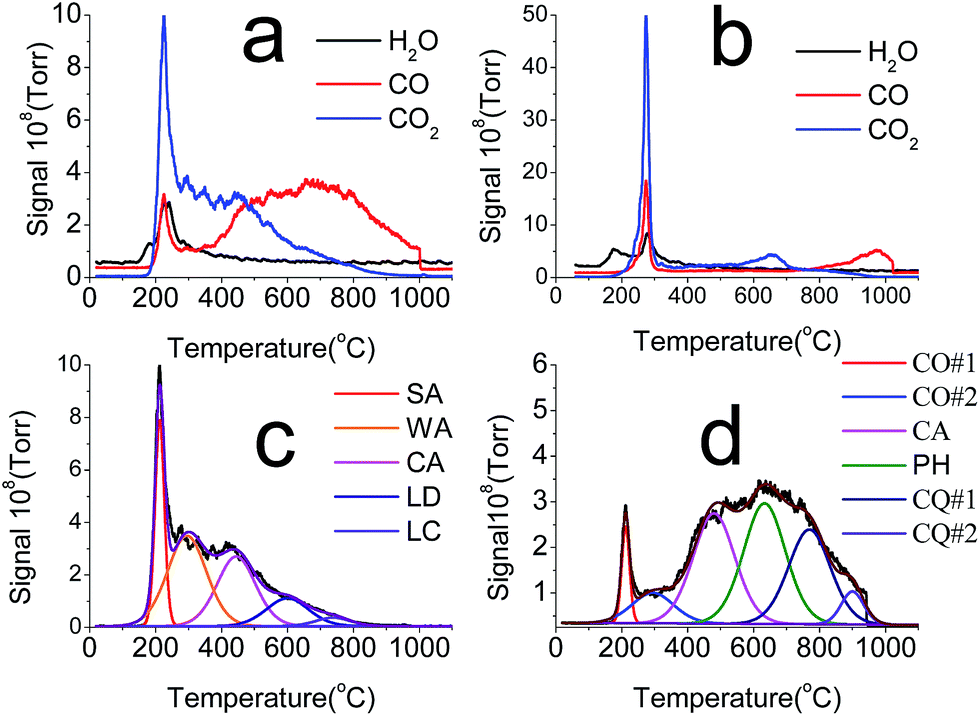 | ||
| Fig. 3 TPD profiles: (a) CO2, CO, and H2O from OAC, (b) CO2, CO, and H2O from GO, (c) deconvolution of TPD-CO2 of OAC, (d) deconvolution of TPD-CO of OAC. | ||
| Sample | Physical H2O mmol g−1 | Formed H2O mmol g−1 | O in CO mmol g−1 | O in CO2 mmol g−1 | Total O (except physical H2O) mmol g−1 |
|---|---|---|---|---|---|
| a The data in parentheses are weight percentage. | |||||
| AC | — | — | 1.2(1.3%) | — | 2.1(4.7%) |
| OAC | 2.3(3.6%)a | 4.0(6.4%) | 5.2(8.3%) | 8.8(14.1%) | 18.0(28.8%) |
| GF | — | — | 0.1(0.0%) | 0.2(0.0%) | 0.3(0.5%) |
| GO | 4.8(8.6%) | 8.0(14.4%) | 5.2(8.4%) | 14.5(23.1%) | 27.7(45.9%) |
The total oxygen contents of the OAC and GO samples increased to 18.0 and 27.7 mmol g−1, respectively. These contents are 7.6 and 91.3 times higher than their original contents. According to the reports in literature, the oxidation method used in this study is likely more effective to introduce OCFGs on AC, and the oxygen content of OAC reached 28.8 wt%, which is quite high in comparison with others.44 It is interesting to note that the total oxygen content in GO is even 54% higher than that of OAC, although the BET surface area of the former is only one-hundredth of the latter. Some studies have used the TPD-MS to characterize the total oxygen content of GO, as reported in literatures.45,46 However, it is difficult to determine the H2O content due to its weak heat stability of GO in the previous studies. The physical water and the water formed during the decomposition of OCFGs were clearly distinguished in this study, as shown in Fig. 3b. The H2O peak formed by the decomposition of OCFGs was observed at approximately 200 °C, as shown in Fig. 3b, which is different from the H2O peak for the physical desorption at approximately 120 °C. In the present study, the oxygen contained in the H2O formed by the decomposition of OCFGs was also included in the total oxygen content of the adsorbent, as shown in Table 3. The oxygen content contributed from the H2O formed at approximately 200 °C can be as high as 14.4% of the total oxygen released. Consequently, this part of oxygen should be considered when estimating the total oxygen concentration and the carboxyl concentration by using the TPD-MS method.
| Sample | SBET (m2 g−1) | qm (mg-N g−1) | K (g μg−1) | R2 | Equilibrium capacity (mg-N g−1) at 300 ppmw |
|---|---|---|---|---|---|
| AC | 2051.5 | 16.7 | 0.020 | 0.994 | 13.9 |
| OAC | 1028.9 | 35.7 | 0.080 | 0.998 | 33.1 |
| GF | 5.9 | 1.9 | 0.005 | 0.992 | 1.1 |
| GO | 9.4 | 29.4 | 0.024 | 0.993 | 24.4 |
The CO2 and CO evolution profiles were further deconvoluted to obtain qualitative and quantitative information about various OCFGs on AC and OAC. This deconvolution method has been widely used for different carbon materials, such as the activated carbon,44 carbon nanofibers,47 carbon nanotubes, and carbon xerogels.48 The TPD-MS method has been proven to be more accurate than FTIR, and XPS, especially for AC.24 The deconvolution results of CO2 and CO for OAC are shown in Fig. 3c and d. The evolution of CO2 mainly results from the decomposition of strongly acidic carboxylic groups (SA) at 240 °C, weak acidic carboxylic groups (WA) at 350 °C, carboxylic anhydrides (CA) at 450 °C, lactones at 620 °C (LD) and other lactones at 760 °C (LC). In the case of CO, the dominant contribution is assigned to CA at 450 °C, phenol (PH) at 710 °C, carbonyls plus quinones (CQ#1, CQ#2) at 900 °C, and pyrones (PY) at 1000 °C.49 It can be seen that different kinds of OCFGs, especially the carboxylic and phenolic groups, were introduced on the AC surface by the APS oxidation modification.
In the case of GO, to the best of our knowledge, the present study is the first time to use TPD-MS for identification of OCFGs on GO. Majority of OGFGs on GO were decomposed to form CO2, CO and H2O at approximately 250 °C. The measured oxygen content that was contributed by the decomposition of OCFGs to form CO and CO2 at approximately 250 °C is approximately 12 mmol g−1, which comprises 43% of the total oxygen contents. Based on the literatures,46 the release of CO and CO2 at this temperature range might be due to the decomposition of epoxides and hydroxyls, although there is still a matter of some debate. The measured oxygen content contributed by the decomposition of OCFGs in other temperature range is relatively small, as shown in Fig. 3.
XRD analysis was also used to investigate the structures of these adsorbents. The XRD patterns of GF, GO and GO after adsorption are presented in Fig. 4. The intensive peak of GF at 2θ = 26.5° matches the interlayer distance of 3.36 Ǻ for GF. This peak vanishes after the oxidative modification. Instead, a broad and relatively weak peak at 2θ = 9.9° appears, which corresponds to the interlayer spacing of 8.9 Ǻ. The results clearly indicate that the oxidative modification enlarged the interlayer spacing of the GF layers due to the formation of different OCFGs, such as hydroxyl, carboxyl or epoxy groups, as also reported by Cote et al.30 In addition, the XRD pattern for GO after quinoline adsorption was also measured. As shown in Fig. 4, the interlayer spacing of GO after quinoline adsorption is 10.1 Ǻ, which is significantly larger than that of GO before adsorption. It is likely that an appreciable fraction on the surface of GO layers is actually accessible to quinoline in the solution. Therefore, it indicates that either the carboxyl on the edge of GO or some OCFGs on the GO interlayer could contribute to the quinoline adsorption.
 | ||
| Fig. 4 XRD patterns of GF, GO and GO after adsorption. | ||
3.3. Adsorption performance of various adsorbents
 | ||
| Fig. 5 Effect of contact time on removal of quinoline in heptane. | ||
The adsorption isotherms for the AC, GF, OAC and GO samples were measured using the MF with quinoline in heptane. The equilibrium isotherms for the adsorption of quinoline on AC, OAC, GF, and GO are shown in Fig. 6. By treatment of the experimental data, it was found that all isotherms fit well the Langmuir adsorption isotherm model. The qm and K, values for adsorption of quinoline on these adsorbents are summarized in Table 3 for easy comparison. In comparison of AC and OAC, the oxidation-modified activated carbons (OAC) has much higher adsorption capacities than AC, as expected. OAC gave the highest quinoline capacity with a qm value of 35.7 mg-N g−1 among the carbon adsorbents tested in this work due to its highest oxygen content. On the other hand, GF has almost no adsorption ability for quinoline. But GO has a much higher capacity for quinoline than original GF. The qm value for GO is 29.4 mg-N g-GO−1, which is considerably higher than those reported before. For example, Cu+Y has been reported as a potential adsorbent to remove nitrogen compounds, but its saturation capacity is only 3 mg-N g-adsorbent−1 at the equilibrium concentration of 83 ppmw.17
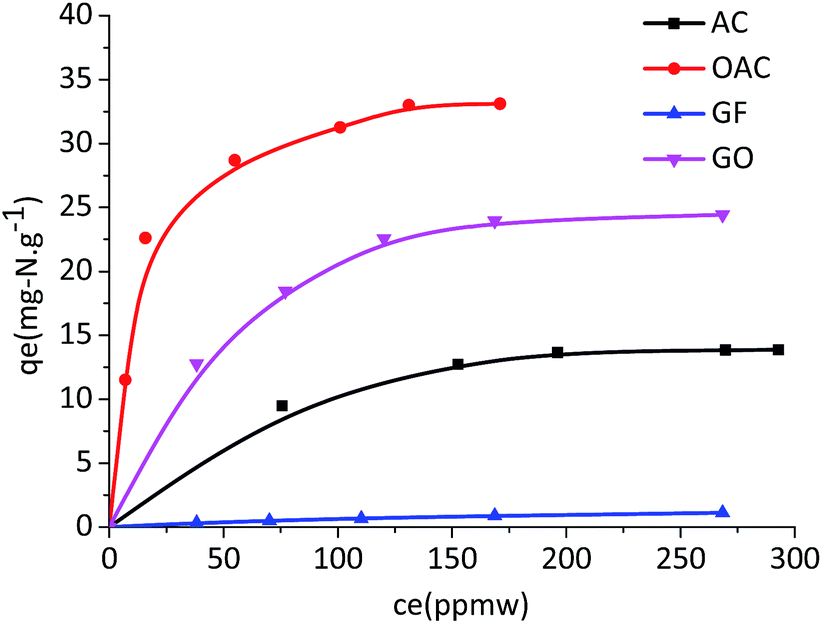 | ||
| Fig. 6 Equilibrium isotherms for the adsorption of quinoline from MF in heptane. | ||
Recently, MOF materials have also studied as adsorbents for nitrogen removal with a maximum capacity of 16 mg-N g-adsorbent−1 at the equilibrium concentration of 300 ppmw.21 A commercial AC with a high surface area and relatively large amount of oxygen content was reported to be 17 mg-N g-adsorbent−1, which is the maximum capacity for adsorption of quinoline among all carbon adsorbents reported in literature.15,19 It is clear that OAC has the highest qm value for quinoline.
In order to examine the effect of the oxygen functional groups on the ADN performance easily, the relationship between the total oxygen content of the adsorbents and their saturate adsorption capacity measured is shown in Fig. 7. There is no good relationship between them. However, when carefully examining the data, it is found that for the activated-carbon-based adsorbents (AC and OAC), the adsorption capacity increases with increasing oxygen content in the adsorbents, while the graphite-based adsorbents also show the same trend with the change of the oxygen content (with similar slope). It indicates that for both cases, the oxygen functional groups on the adsorbents play a critical role in determining their adsorption capacity. The significant higher adsorption capacity of the activated-carbon-based adsorbents than that of the graphite-based adsorbents at the same oxygen content may be contributed to their difference in the textural structure. GO has relative small interlayer space (0.89 nm), which means part of the oxygen groups on the basal plane between two layers may be limited for the approach of quinoline molecule. In addition, it should be mentioned that the GO could not be dispersed in hexane (the solvent in our MF) even after sonication due to its weak polarity.27,51,52 In other word, the accessibility of the species to these oxygen functional groups might be the key for it. The further investigation is necessary to clarify it. On the other hand, GF, which has nearly no pore structure and no oxygen groups, exhibited a low, but certain, capacity for quinoline, indicating that the adsorption of quinoline may also be conducted through the π–π interaction with the basal graphene surface, but such interaction is relative weaker than the acid–base interactions and H-bonding interactions, as confirmed by the K value (0.005 g μg−1) of GF in comparison with that (0.024 g μg−1) of GO. In the case on AC, the contribution to the capacity for quinoline may be mainly due to the π–π interactions between the aromatic rings in quinoline and basal graphite plane in AC.16,19 The increase in the capacity of OAC for quinoline in comparison with AC is likely primarily due to the increase in its total concentration of oxygen groups on the surface, which interact with the quinoline molecules dominantly through the acid–base interaction and H-bonding interaction.16 The reasons for the dramatic higher capacity of GO than that of GF may be both the higher concentration of oxygen groups and the larger interlayer space of the former than those of the latter.
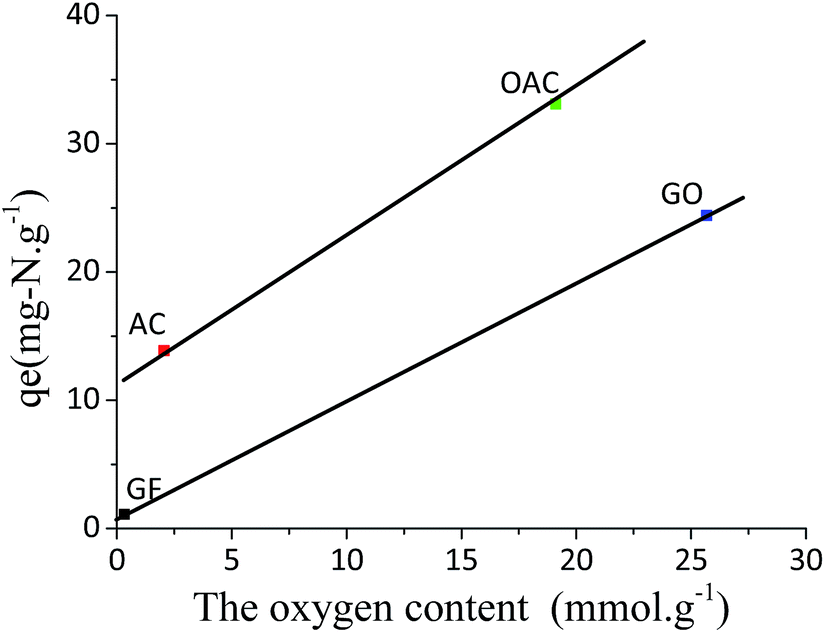 | ||
| Fig. 7 Correlation between the total oxygen content and the quinoline adsorption capacity at 300 ppmw. | ||
In summary, the multiple mechanisms work together to determine the adsorption performances of the carbon-based adsorbents in removing the nitrogen compounds from liquid hydrocarbon streams. The oxygen groups are very important for enhancing the adsorption capacity of the carbon adsorbents for quinolone in comparison with other factors.
3.4. Regeneration of spent adsorbent
Some researchers have studied the regeneration of the spent AC adsorbents, and found that about 70% adsorption capacity of the adsorbent can be recovered after the solvent extraction in toluene.6,42 However, there has no report about the regenerability of GO. The adsorption capacity of the spent GO, regenerated by using different solvents decreased in the solvent order of DMF (92%) > acetone (46%) > ethanol (35%) > toluene (25%), which is consistent with their polarity: DMF (6.4) > acetone (5.4) > ethanol (4.3) > toluene (2.4), indicating that the polarity of the solvents is important for effective regeneration. The temporary exploration in the regeneration of the spent GO shows that the majority of the capacity of GO can be recovered by the ultrasonic extraction, as the majority of the adsorptions on GO is reversible.4. Conclusions
The adsorption behaviours of the four carbon-based adsorbents (AC, OAC, GF and GO) with different textural structures and concentrations of various oxygen functional groups were studied for removing quinoline from liquid hydrocarbons. The adsorption capacity (35.7 mg-N g−1) of OAC prepared in this work is much higher than those reported in literature due to its higher concentrations of the oxygen functional groups. GO prepared from the oxidation of GF has also shown much higher adsorption capacity (29.4 mg-N g−1) for quinoline. Different types of the oxygen groups might play different roles in determining the adsorption performance. The carboxyl and phenol group might play a crucial role in determining the adsorption performance of the carbon-based adsorbent for selective removal of quinoline from liquid hydrocarbon streams and the accessibility of these groups on the surface is another key factor to affect the adsorption performance.Acknowledgements
The financial support from the National Nature Science Foundation of China (No. 21306121), the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions and the China Postdoctoral Science Foundation (No. 2013M541723) are gratefully acknowledged.Notes and references
- U. T. Turaga, X. L. Ma and C. S. Song, Catal. Today, 2003, 86, 265–275 CrossRef CAS.
- Y. Sano, K.-H. Choi, Y. Korai and I. Mochida, Appl. Catal., B, 2004, 53, 169–174 CrossRef CAS PubMed.
- H. Yang, J. Chen, C. Fairbridge, Y. Briker, Y. J. Zhu and Z. Ring, Fuel Process. Technol., 2004, 85, 1415–1429 CrossRef CAS PubMed.
- S. Achmann, G. Hagen, M. Haemmerle, I. M. Malkowsky, C. Kiener and R. Moos, Chem. Eng. Technol., 2010, 33, 275–280 CrossRef CAS PubMed.
- B. van de Voorde, M. Boulhout, F. Vermoortele, P. Horcajada, D. Cunha, J. S. Lee, J.-S. Chang, E. Gibson, M. Daturi and J.-C. Lavalley, J. Am. Chem. Soc., 2013, 135, 9849–9856 CrossRef CAS PubMed.
- Y. Sano, K.-H. Choi, Y. Korai and I. Mochida, Appl. Catal., B, 2004, 49, 219–225 CrossRef CAS PubMed.
- P. Zeuthen, K. G. Knudsen and D. D. Whitehurst, Catal. Today, 2001, 65, 307–314 CrossRef CAS.
- M. Egorova and R. Prins, J. Catal., 2004, 221, 11–19 CrossRef CAS.
- Y. Sano, K.-H. Choi, Y. Korai and I. Mochida, Energy Fuels, 2004, 18, 644–651 CrossRef CAS.
- K.-H. Choi, Y. Korai, I. Mochida, J.-W. Ryu and W. Min, Appl. Catal., B, 2004, 50, 9–16 CrossRef CAS PubMed.
- M. Egorova and R. Prins, J. Catal., 2004, 224, 278–287 CrossRef CAS PubMed.
- R. Wandas and T. Chrapek, Fuel Process. Technol., 2004, 85, 1333–1343 CrossRef CAS PubMed.
- R. Prins, M. Egorova, A. Röthlisberger, Y. Zhao, N. Sivasankar and P. Kukula, Catal. Today, 2006, 111, 84–93 CrossRef CAS PubMed.
- M. Almarri, X. L. Ma and C. S. Song, Ind. Eng. Chem. Res., 2008, 48, 951–960 CrossRef.
- M. Almarri, X. L. Ma and C. S. Song, Energy Fuels, 2009, 23, 3940–3947 CrossRef CAS.
- J. H. Kim, X. L. Ma, A. Zhou and C. S. Song, Catal. Today, 2006, 111, 74–83 CrossRef CAS PubMed.
- A. J. Hernández-Maldonado and R. T. Yang, Angew. Chem., 2004, 116, 1022–1024 CrossRef PubMed.
- A. Zhou, X. L. Ma and C. S. Song, J. Phys. Chem. B, 2006, 110, 4699–4707 CrossRef CAS PubMed.
- J. Wen, X. Han, H. Lin, Y. Zheng and W. Chu, Chem. Eng. J., 2010, 164, 29–36 CrossRef CAS PubMed.
- L.-L. Xie, A. Favre-Reguillon, X.-X. Wang, X. Fu and M. Lemaire, J. Chem. Eng. Data, 2010, 55, 4849–4853 CrossRef CAS.
- M. Maes, M. Trekels, M. Boulhout, S. Schouteden, F. Vermoortele, L. Alaerts, D. Heurtaux, Y. K. Seo, Y. K. Wang and J. S. Chang, Angew. Chem., 2011, 123, 4296–4300 CrossRef PubMed.
- J. Lemus, C. M. Neves, C. F. Marques, M. G. Freire, J. A. Coutinho and J. Palomar, Environ. Sci.: Processes Impacts, 2013, 15, 1752–1759 CAS.
- M. Seredych, J. Lison, U. Jans and T. J. Bandosz, Carbon, 2009, 47, 2491–2500 CrossRef CAS PubMed.
- N. Li, J. Zhu, X. L. Ma, Q. Zha and C. S. Song, AIChE J., 2013, 59, 1236–1244 CrossRef CAS PubMed.
- R. Paul, T. Voskuilen, D. Zemlyanov, T. L. Pourpoint and T. S. Fisher, ASME 2012 International Mechanical Engineering Congress and Exposition, Houston, 2012 Search PubMed.
- C. Nethravathi and M. Rajamathi, Carbon, 2008, 46, 1994–1998 CrossRef CAS PubMed.
- F. Perreault, A. F. de Faria and M. Elimelech, Chem. Soc. Rev., 2015, 44(16), 5861–5896 RSC.
- H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS PubMed.
- W. Chen, L. Yan and P. R. Bangal, Carbon, 2010, 48, 1146–1152 CrossRef CAS PubMed.
- L. J. Cote, F. Kim and J. Huang, J. Am. Chem. Soc., 2008, 131, 1043–1049 CrossRef PubMed.
- H. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53–56 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- M. Barathi, A. S. K. Kumar, C. U. Kumar and N. Rajesh, RSC Adv., 2014, 4, 53711–53721 RSC.
- S.-M. Hong and K. Lee, RSC Adv., 2014, 4, 56707–56712 RSC.
- R. Sitko, E. Turek, B. Zawisza, E. Malicka, E. Talik, J. Heimann, A. Gagor, B. Feist and R. Wrzalik, Dalton Trans., 2013, 42, 5682–5689 RSC.
- X. Ren, J. Li, X. Tan and X. Wang, Dalton Trans., 2013, 42, 5266–5274 RSC.
- N. Li, X. L. Ma, Q. F. Zha and C. S. Song, Energy Fuels, 2010, 24, 5539–5547 CrossRef CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- N. Mahata, M. Pereira, F. Suárez-García, A. Martínez-Alonso, J. Tascón and J. Figueiredo, J. Colloid Interface Sci., 2008, 324, 150–155 CrossRef CAS PubMed.
- A. Srivastav and V. C. Srivastava, J. Hazard. Mater., 2009, 170, 1133–1140 CrossRef CAS PubMed.
- N. A. Khan, J. W. Jun, J. H. Jeong and S. H. Jhung, Chem. Commun., 2011, 47, 1306–1308 RSC.
- X. Han, H. Lin and Y. Zheng, Chem. Eng. J., 2014, 243, 315–325 CrossRef CAS PubMed.
- W. Li, H. Tang, Q. Liu, J. Xing, Q. Li, D. Wang, M. Yang, X. Li and H. Liu, Biochem. Eng. J., 2009, 44, 297–301 CrossRef CAS PubMed.
- N. Li, X. L. Ma, Q. Zha, K. Kim, Y. Chen and C. S. Song, Carbon, 2011, 49, 5002–5013 CrossRef CAS PubMed.
- P. Solís-Fernández, R. Rozada, J. I. Paredes, S. Villar-Rodil, M. J. Fernández-Merino, L. Guardia, A. Martínez-Alonso and J. M. D. Tascón, J. Alloys Compd., 2012, 536, S532–S537 CrossRef PubMed.
- L. M. Pastrana-Martínez, S. Morales-Torres, V. Likodimos, P. Falaras, J. L. Figueiredo, J. L. Faria and A. M. Silva, Appl. Catal., B, 2014, 158, 329–340 CrossRef PubMed.
- J.-H. Zhou, Z.-J. Sui, J. Zhu, P. Li, D. Chen, Y.-C. Dai and W.-K. Yuan, Carbon, 2007, 45, 785–796 CrossRef CAS PubMed.
- H. F. Gorgulho, J. P. Mesquita, F. Gonçalves, M. F. R. Pereira and J. L. Figueiredo, Carbon, 2008, 46, 1544–1555 CrossRef CAS PubMed.
- J. L. Figueiredo and M. F. R. Pereira, Catal. Today, 2010, 150, 2–7 CrossRef CAS PubMed.
- Y. S. Bae, M. B. Kim, H. J. Lee, C. H. Lee and J. Wook Ryu, AIChE J., 2006, 52, 510–521 CrossRef CAS PubMed.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463–470 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed physical and chemical properties of four materials. See DOI: 10.1039/c5ra09228k |
| This journal is © The Royal Society of Chemistry 2015 |