
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functional wound dressing materials with highly tunable drug release properties†
Tina
Maver
a,
Silvo
Hribernik
a,
Tamilselvan
Mohan
*b,
Dragica Maja
Smrke
c,
Uroš
Maver
*de and
Karin
Stana-Kleinschek
*a
aFaculty of Mechanical Engineering, Laboratory for Characterisation and Processing of Polymers, University of Maribor, Smetanova 17, SI-2000 Maribor, Slovenia. E-mail: karin.stana@um.si; Fax: +386 2 220 7990; Tel: +386 2 220 7881
bUniversity of Graz, Institute for Chemistry, Heinrichstrasse 28, 8010 Graz, Austria. E-mail: tamilselvan.mohan@uni-graz.at; Tel: +43 316 380 5413
cUniversity Medical Centre Ljubljana, Zaloška cesta 2, SI-1000, Ljubljana, Slovenia
dFaculty of Medicine, Department of Pharmacology and Experimental Toxicology, University of Maribor, Taborska ulica 8, SI-2000 Maribor, Slovenia. E-mail: uros.maver@um.si; Fax: +386 2 234 5923; Tel: +386 2 234 5823
eInstitute of Palliative Medicine and Palliative Care, Faculty of Medicine, University of Maribor, 2000 Maribor, Slovenia
First published on 9th September 2015
Abstract
Wound dressings, capable of local controlled delivery of non-steroid anti-inflammatory pain-killing drugs (NSAIDs) to the wound bed, offer great potential to accelerate wound healing, hence increase the quality of patient life. With local NSAID delivery, unwanted side effects encountered in their systemic delivery, are drastically diminished. In this study, four functional fibrous wound dressing materials, namely viscose, alginate, sodium carboxymethyl cellulose (Na-CMC) and polyethylene terephthalate (PET) loaded with a NSAID, diclofenac sodium (DCF) are prepared, and their suitability to tune the release rate of DCF is evaluated. Through careful examination of material–drug combinations, in terms of their physicochemical properties (air permeability, wettability and water retention) and structural/morphological properties (infrared spectroscopy, wide angle X-ray scattering and scanning electron microscopy), possible wound care applications are proposed. In vitro release studies using an automated Franz diffusion cell system, combined with UV-Vis absorption spectroscopy for drug release profile determination, are performed as the final pre-formulation test. Results showed significant differences in the release profiles between different material–drug combinations, making the examined materials highly applicable for several wound care applications. The present study presents a novel cost effective approach for preparation of drug loaded wound dressing materials without a sacrifice in patient safety. Additionally, novel methods and material–drug combinations are introduced, paving the way for possible future wound treatment options.
1. Introduction
A wound commonly refers to a skin injury caused by trauma or surgery. Wound healing generally follows a well-defined yet complex cascade of processes commonly divided into four main stages; coagulation, inflammation, cell proliferation with repair of the matrix, and epithelialization with remodeling of the scarred tissue.1 Depending on the extent and depth of skin damage, the entire healing process can last for several months.2 Among the most important conditions affecting wound healing start, are the wound cleanliness, a suitable blood supply, and the absence of necrotic leftovers and fibrin plaques.3 Once the healing is underway, an appropriate moisture balance and prevention of infections, as well as exudate management, are necessary to assure effective healing.4 Although multi-layered and multi-functional wound dressings are not a novelty in wound care/healing process,5 existing products do not address the challenging issues in wound treatment, such as controlled therapeutic action or wound type specific healing.Different fiber forming polymers are often used as drug carriers, absorbents or as moisturizers in wound dressings.6 Their semi-crystalline structures, nano-, micro- and especially macro-porosity allow them to incorporate, bind and release different amounts of active ingredients according to their respective structural and physico-chemical properties.7 The most commonly used are PET, cellulose and its derivatives and regenerated cellulose such as viscose, as well as other types of polysaccharides. PET is a hydrophobic polymer, which is often used as an inert layer of the dressing, suitable for contact with the damaged skin.8 Regenerated cellulose (viscose) and cellulose derivatives (sodium salt of carboxymethyl cellulose, Na-CMC), as well as alginate, are among the most common functional parts of different modern wound dressings.9 Their final formulation shapes range from fibers, non-woven materials and hydrogels to foams.8
Lately, novel advanced wound dressing formulations are explored in order to achieve controlled release and delivery of drugs to wound sites.10,11 The release from such materials is sparsely reported in literature with few clinical studies carried out to date.12,13 Especially little or almost no literature is available regarding the controlled wound type specific drug delivery using polymeric drug loaded materials as dressings. Despite the well-known fact that different wounds exhibit significantly altered wound bed conditions,14 they provide different physiological conditions for drug release. Polymer-based dressings employed for controlled drug delivery to wounds include hydrogels such as poly(lactide-co-glycolide),15 poly(vinyl pyrrolidone),16 poly(vinyl alcohol)17 and poly(hydroxylalkyl-methacrylates),18 polyurethane-foam,19 hydrocolloid20 and alginate dressings.21 Other polymeric dressings reported in literature for this purpose include novel hybrid formulations prepared from hyaluronic acid,22 collagen23 and chitosan.24,25
Drug release from polymeric formulations is mostly controlled by one or more physical processes including (a) hydration of the polymer, (b) swelling and gel formation, (c) diffusion of the drug through the matrix and (d) eventual erosion of the matrix.26 Since wounds exhibit different extents of exudation, it is expected that wound specific healing can be achieved by combining swelling, erosion and subsequent drug diffusion kinetics as part of the controlled drug release mechanism. In fact, most of the recently researched materials intended for wound dressings (either natural or synthetic) release incorporated drugs by a combined mechanism of either two or three above mentioned principles.27,28
Non-steroid anti-inflammatory drugs (NSAIDs) are important drugs in relieving pain, fighting fever and decreasing inflammation. However, both NSAIDs and their selective cyclooxygenase-2 (COX-2) inhibitors inhibit PGE2 production, which might exacerbate excessive scar formation, especially when used during the later proliferative phase. Nevertheless, it was shown that pain reduction induced decrease in stress, can positively affect wound healing, resulting in shortened healing times.29 Based on scientific and clinical evidence, pain can significantly slow down the healing process (mostly through stress induced release of hormones like cortisol and norepinephrine), which results in decreased patient quality of life as well as in exponentially increased personal and public expenditures.30–32 Mostly NSAIDs are taken through systemic administration (i.e., in the form of pills), whereas several approaches have been and are researched towards their integration into different wound dressing formulations.33–36 Although antibiotics are not the preferred type of drugs for local treatment due to possible resistance acquisition of commensal bacteria, there are several interesting studies available about preparation of dressing, combining NSAIDs and antimicrobials.37–39
The purpose of this study was therefore to prepare wound dressing materials with incorporated NSAIDs and to study their efficiency related to material performance and drug release. For this purpose, only commercially available and clinically approved materials were used. Four wound dressing materials, widely differing in their ability to take up liquids, namely sodium salt of carboxymethyl cellulose (Na-CMC), alginate (ALG), viscose and polyethylene terephthalate (PET) were chosen and subsequently integrated or loaded with the (NSAID), diclofenac (DCF). The structural and surface properties of the material–DCF potential wound dressing combinations were analyzed in detail using contact angle, water retention and air permeability measurements, wide angle X-ray diffraction (WAXS) and scanning electron microscopy (SEM). The application potential of the chosen materials was shown in the release of the NSAID DCF in vitro using Franz diffusion cell release studies followed by quantification using UV-Vis absorption spectroscopy. Through preparation of four different material–DCF combinations and their ability to release DCF, this study was intended to assess the suitability of different wound dressing materials for effective and safe pain reduction in relation to the treatment of different wound types. To our best knowledge, WAXS was never before used in examination of novel wound dressing formulations. Our study aimed at providing a novel approach towards preparation of cost effective drug loaded wound dressing solutions without the sacrifice of patient safety. It also provides a possible novel PET based wound dressing with potential for future wound care applications.
2. Materials and methods
2.1. Materials
Four commercially available materials differing in their surface properties (wettability and composition) were used as wound dressings. Viscose nonwoven (specific surface area mass = 175 g m−2) was purchased from KEMEX, Netherlands. Fibrous alginate (ALG) and carboxymethyl cellulose (Na-CMC; commercial, clinically used, wound dressing Aquacel®) nonwoven were purchased from Sigma-Aldrich, Slovenia and ConvaTec, USA, respectively. Polyethylene terephthalate, PET (100%, specific surface area = 75 g m−2) in the form of a mesh (mesh size of 0.8 mm) was purchased from BETI, Slovenia. Diclofenac sodium (DCF) was purchased from Sigma-Aldrich, Germany. All the materials were used as received without any further modification prior to sample preparation or testing. Ultra-pure water (18.2 MΩ cm at 25 °C) from an ELGA PureLab water purification system (Veolia Water Technologies, UK) was used for preparation of all solutions.2.2. Integration of drug molecules into wound dressing materials
The wound dressing samples were cut into 1 × 1 cm squares and impregnated with DCF (dissolved in ultra-pure water, 1 mg ml−1) for 15 minutes. Afterwards, the samples were dried in an oven at 50 °C for 5 minutes, cooled down to room temperature and finally flushed with nitrogen gas. The as-prepared samples were immediately used for in vitro release testing and characterization.2.3. Methods
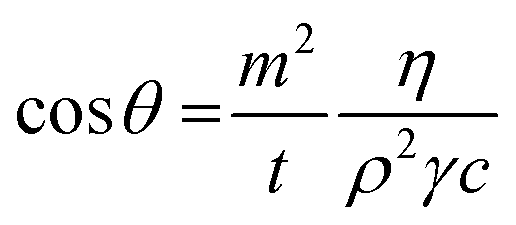 | (1) |
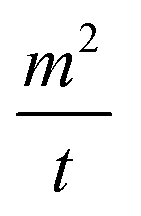 is the capillary velocity, η is the liquid viscosity, ρ is the liquid density, γ is the surface tension of the liquid and c is a material constant.4,5,40
is the capillary velocity, η is the liquid viscosity, ρ is the liquid density, γ is the surface tension of the liquid and c is a material constant.4,5,40
All measurements were performed on three independent samples at three different sample regions. An average value was calculated and the standard error is reported.
The reference air permeability was calculated using the following eqn (2):
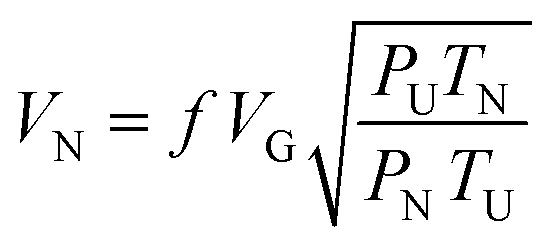 | (2) |
Measurements were performed using the Louis Schopper apparatus (Leipzig, Austria). The thickness of the pressure plate and its surface area were 4 mm and 1000 mm2 (with a diameter of 35.68 mm), respectively. For each sample five measurements were performed and an average value was calculated with an added standard error.
The withdrawn sample volumes were replaced by fresh ultra-pure water. Due to sample withdrawal, followed by sample dilution through media replacement, sink conditions were assured. In calculation of concentrations using the Beer–Lambert Law, this dilution was accounted for. All release studies were performed in three parallels. For the determination of final incorporated DCF amount, pieces of the same size (1 × 1 cm squares) of each material with incorporated drug were shredded and immersed into 20 ml absolute ethanol. After 48 h of shaking, DCF concentration was determined by UV-Vis spectrophotometry. Furthermore, to confirm the complete release of DCF the samples were dried in oven at 50 °C for 15 minutes and analyzed by ATR-IR.
To compare the differences in release rates of wound dressing materials loaded with DCF drug, a regression analysis was performed using the GraphPad Prism Software Version 5.01. A difference is considered to be significant, when the obtained p-value is lower than 0.05 (p < 0.05). The calculated p-values are given in the ESI (Table S1†).
3. Results and discussion
3.1. Wettability, water retention and moisture content of unloaded samples
Keeping the wound's surface moist is the fundamental principle of open wound treatment.41 Several methods can be applied to evaluate a material capability to assure appropriate moisture in the wound bed. The common denominator of such measurements is hydrophilicity, which is connected with the water contact angle and is reflected in the water retention value, as well as in the materials moisture content. The water contact angles (CA(H2O)) and water retention values of unloaded wound dressing samples are shown in Fig. 1.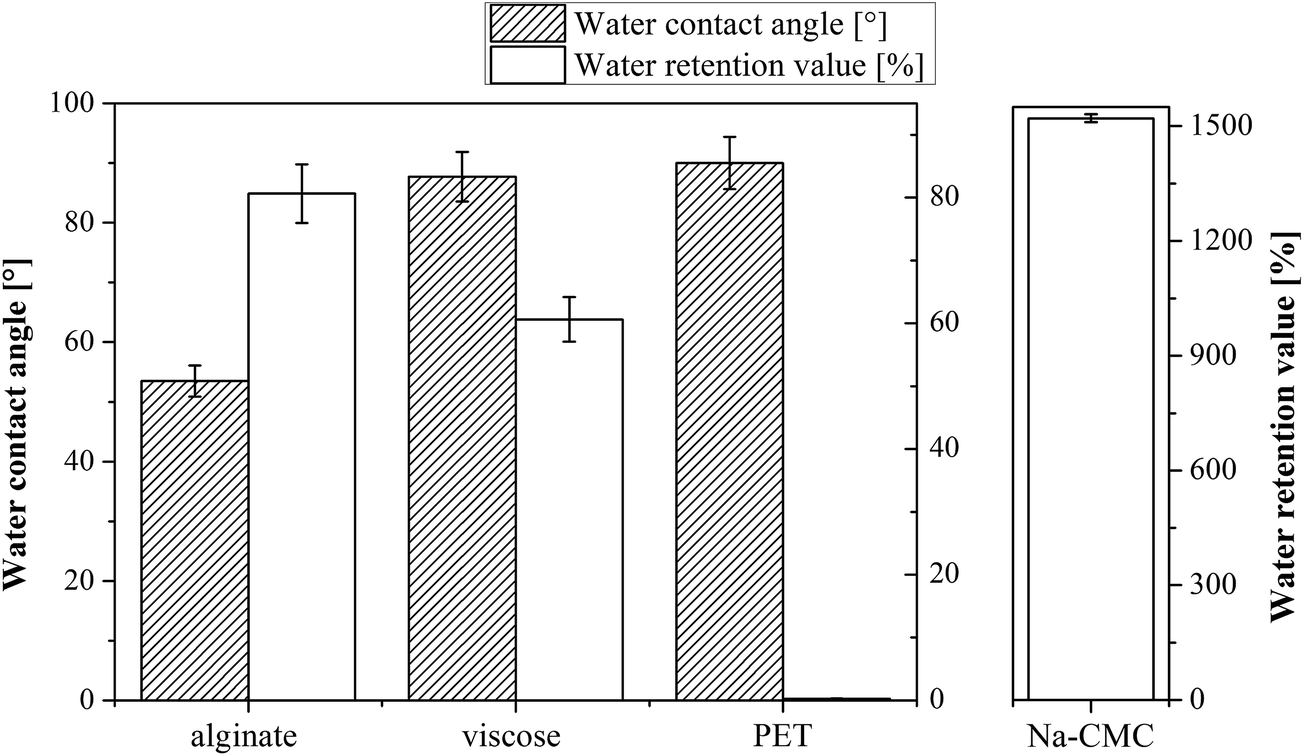 | ||
| Fig. 1 (A) The water contact angle and water retention properties of the wound dressing materials prior to drug incorporation. PET water retention value is not missing, but almost zero (0.3%), and therefore cannot be seen in the diagram. (B) Na-CMC water retention value is shown in a separate diagram due to its much higher value, when compared to the other samples, shown in part A of the diagram. Water contact angle for Na-CMC (Aquacel®) could not be obtained. | ||
It is expected that CA(H2O) are inversely proportional to the water retention values – the larger the contact angle, the lower the water retention value, but while measuring the mentioned on wound dressing materials, their 3D structure (often fibrous), in particular in the case of PET and viscose materials, plays an important role in regulating their overall hydrophilicity. The relation between CA(H2O) and water retention values is therefore not straightforward, as shown also in case of our results. The CA(H2O) values demonstrate that the most hydrophilic materials are clearly Na-CMC and alginate compared to other two materials (viscose and PET). Alginate exhibits the lowest CA(H2O): 54 ± 3°, whereas the measurement of the CA(H2O) is not possible for Na-CMC (Aquacel®) due to its extremely high soaking ability and hydrophilic nature, which is facilitated by a higher content of carboxylic groups. The latter prevented us to determine the capillary velocity, and hence the CA(H2O) value for this sample. From literature it is clear that the contact angle of Na-CMC is very dependent on the material type. A range of CA(H2O) values from low to high can be found in the literature.42,43 Since we are not able to measure this value, probably due to Na-CMC super-molecular structure that allows the material to form a gel-like structure, which results in an extremely high water uptake, this value is missing in Fig. 1. The PET sample, as a known hydrophobic synthetic polymer, exhibited a CA(H2O) of 90 ± 4°.43 Viscose (cellulose, regarded as a hydrophilic material) rather shows a higher CA(H2O): 88 ± 4°, similar to PET. This increased CA(H2O) can be attributed to complex structural properties of cellulose fibers, such as two-phase regime composed of disordered accessible regions and ordered crystalline regions, porous system with voids and interfibrillar interstices, fiber orientation, etc.44,45
By far the highest water retention value was determined for the Na-CMC sample. Indeed, the measured value is so high (1521%) that we have to present it in a separate diagram (Fig. 2B) to allow for a clearer comparison of the other samples. The higher water retention values of Na-CMC can be related to incorporation of a larger number of water molecules by its ionisable carboxylic groups (–COO−). PET samples, on the other hand, exhibit the lowest values for water retention due to their high hydrophobic character. The water is repelled from its surface, and consequently, the resulting value can almost not be seen in Fig. 1 (0.3%). Alginate and viscose exhibit water retention values of 81% and 61%, respectively. These values are in agreement with the values reported in literature for alginate46 and viscose.4 The differences in their water retention properties can be attributed to their functional groups and structural properties (porosity, degree of crystallinity and fiber orientation).45,47
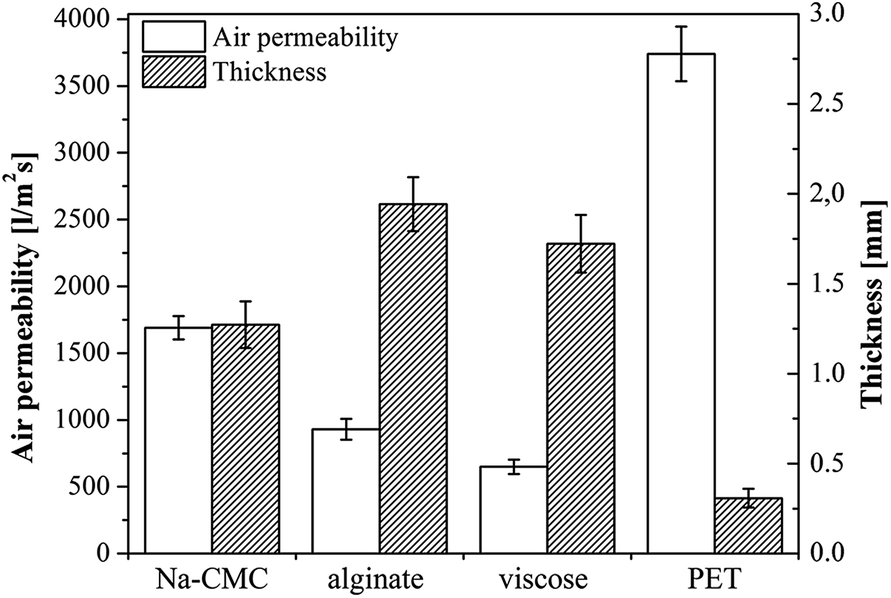 | ||
| Fig. 2 Air permeability and thickness of the unloaded samples. | ||
Moisture content measurement is an important feature, affecting our choice of drug host materials in wound treatment, since it affects the final capacity to uptake fluids when applied on the wound. On the other hand, the initial moisture content can seriously influence the controlled release behavior due to porosity and changes in mechanical strength of the host material. It also affects the release and distribution of drug molecules, especially considering the drug molecules accessibility by the release media.48 More related discussion will be presented in the section explaining drug release studies. The results of materials' moisture content evaluation show a similar order among tested samples as for the CA(H2O), except for Na-CMC, for which CA(H2O) could not be obtained. Na-CMC was initially in a dry form than alginate, but exhibits in its pure form (out of the secondary packaging) a higher moisture content than viscose and PET (Table S1†). Based on the moisture content differences among the used materials, extreme care is necessary during all other testing procedures in order to avoid possible fluctuations in moisture during experimentation. Between measurements, all materials were always placed into tightly sealed Petri dishes and additionally sealed with parafilm strips.
3.2. Air permeability vs. thickness of samples
A medical dressing must be permeable for gasses, but an excessive air permeability, especially a higher moisture vapor transmission rate, could dry out the wound and have a negative effect on healing. The latter is due to two mechanisms, either through direct impact on newly formed cells (excessive drying of the wound bed causes cell death), or by resulting in growing of the dressing into the wound.4 The latter being a result of dressing interaction with newly formed tissue, forming crusts and resulting in an unfavorable effect on the wound's healing rate.49 Given the conditions of the testing method, air permeability is mostly affected by the manufacturing method of the material (knitting and preparation of non-woven), yet sample structure, chemical nature, as well as the samples' thickness must be taken into account. Air permeability of the chosen materials was evaluated against the sample flat thickness. The results are shown in Fig. 2.The expected correlation between thickness and air permeability, namely the thicker the material, the lower is the air permeability, is obtained for Na-CMC and alginate. One could argue that also viscose exhibits the same correlation, especially with the error bars counted in. Based on these results, it is clear that the demonstrated air permeability of Na-CMC, alginate and viscose are satisfactory for ensuring and maintaining optimal wound healing conditions. On contrary, the results also clearly show an extremely high air permeability for PET, not suitable for desired maintenance of a moist wound environment. PET is therefore only applicable either in thicker layers or by preparation of multi-layered wound dressings comprising additional materials.
3.3. DCF incorporation and characterization: ATR-IR and WAXS spectroscopy of samples
ATR-IR spectroscopy is employed to access the chemical composition and structural properties of the unloaded and DCF loaded wound dressing materials. A clear indication of successful DCF incorporation in all samples (Na-CMC, alginate and viscose) except in the case of PET can be seen from the IR spectra shown in Fig. 3. For respective samples, these are organized in a way allowing clear comparison of peaks that can be assigned to DCF. The most important peaks are colored in green. A broad peak between 3700–3000 cm−1 that corresponds to OH vibration can be observed for all unloaded polysaccharide based materials (Na-CMC, alginate and viscose). Pure PET, in contrast, can be characterized by –CH vibrations at 2900 cm−1 and a typical finger print region (650–1700 cm−1).50 After DCF doping the emergence of several new peaks can be observed. Peaks that can be assigned to C–Cl stretching vibrations are visible in the region of 650–780 cm−1, while a band corresponding to CH–N–CH bending vibration can be observed at 1376 cm−1. At 1577 cm−1, R = C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration can be observed. Additional peaks corresponding to R–CO stretching and CH2 bending are visible at 1305 and 1462 cm−1. All these peaks are clearly visible for all DCF loaded polysaccharide-based samples. This is an evidence that DCF is successfully loaded into the used wound dressing materials regardless of the difference in their chemical functionality and structural properties. Even though no peaks corresponding to DCF are observed in the IR spectra for the PET sample, the presence of DCF is clearly evident from the in vitro release results (see Section 3.4) from this sample. A plausible reason can be that the concentration of loaded DCF is too low to be detected by IR.
O stretching vibration can be observed. Additional peaks corresponding to R–CO stretching and CH2 bending are visible at 1305 and 1462 cm−1. All these peaks are clearly visible for all DCF loaded polysaccharide-based samples. This is an evidence that DCF is successfully loaded into the used wound dressing materials regardless of the difference in their chemical functionality and structural properties. Even though no peaks corresponding to DCF are observed in the IR spectra for the PET sample, the presence of DCF is clearly evident from the in vitro release results (see Section 3.4) from this sample. A plausible reason can be that the concentration of loaded DCF is too low to be detected by IR.
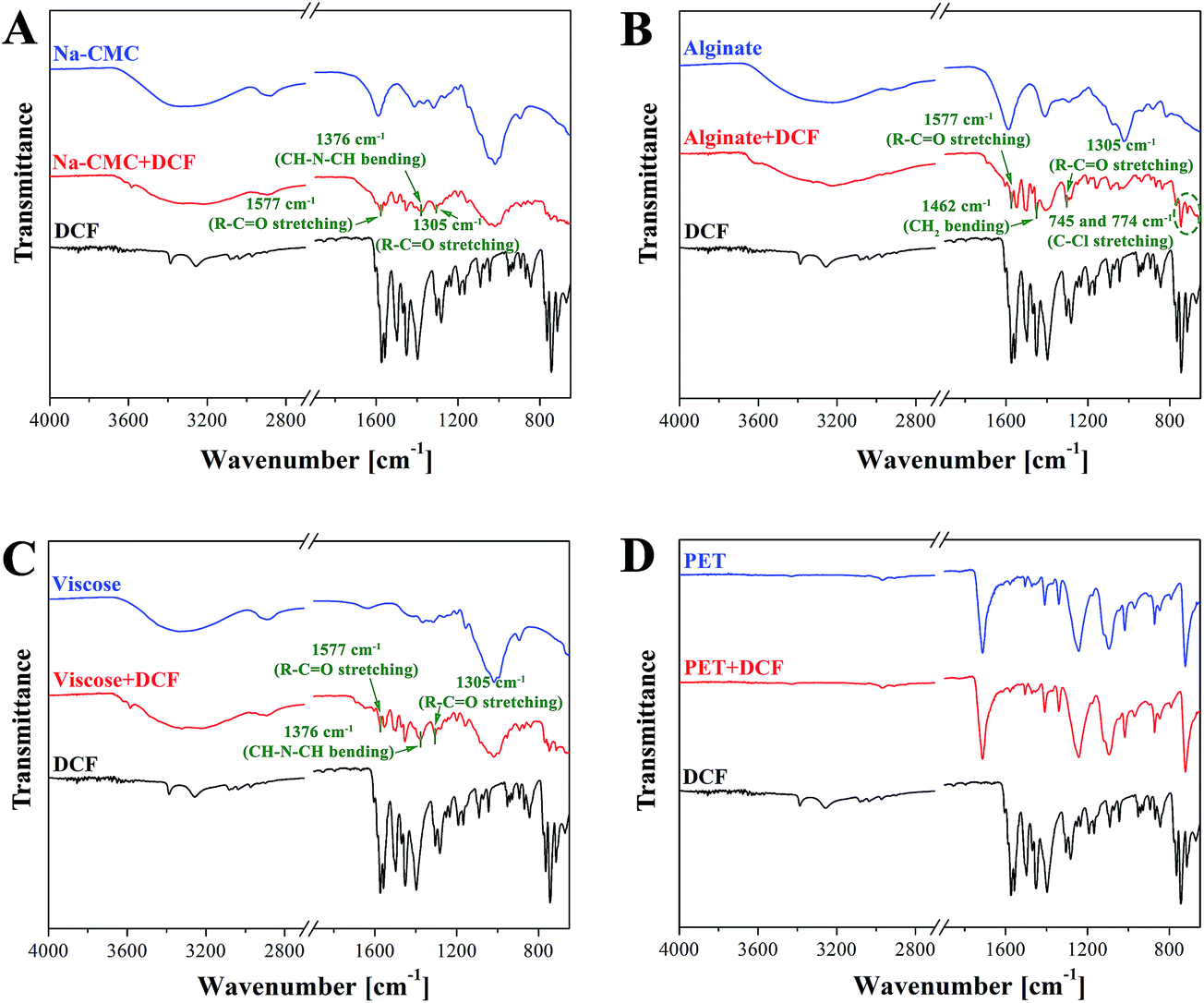 | ||
| Fig. 3 ATR – IR spectra of (A) unloaded Na-CMC, DCF and Na-CMC with incorporated DCF, (B) unloaded alginate, DCF and alginate with incorporated DCF, (C) unloaded viscose, DCF, viscose with incorporated DCF and (D) unloaded PET, DCF and PET with incorporated DCF. | ||
WAXS measurements, as an additional tool to support the findings of ATR-IR, is performed to determine the structure of wound dressing materials as well as to detect the presence of DCF in the samples (Fig. 4). From the obtained diffractograms a DCF corresponding peak around 21°, could be identified for all samples, although the latter is not that evident for the PET sample with the lowest amount of incorporated DCF. This data fits well with ATR-IR results (see Fig. 3d), where almost no peaks related to DCF presence were noted. Apart from the mentioned peak an additional peak around 26° could be observed for the viscose and alginate DCF loaded samples, while the peak around 23°, also assigned to DCF, could be observed for Na-CMC. Although WAXS is not a very frequently used method for this purpose, it still has a higher potential for identification purposes as turned out also in our case. Another useful information we could obtain from the measurements is an indication about crystallinity of the sample. To confirm the overall sample crystallinity a more thorough WAXS analysis would be necessary, but nevertheless, these results suggest that fractions of all samples are amorphous and since the DCF assigned peak around 21° is evident even after incorporation, the drug seems to remain in its crystal form. Most of the diffractograms exhibit broad peaks, a characteristic contribution of the, structural components, lacking order. At this point, we have to stress that the characterization was done at a limited range of angles, therefore an explicit judgement is not possible. Since the crystal structure can significantly affect the materials wetting properties,51 WAXS results will be integrated also in the explanation of the drug release later in the article.
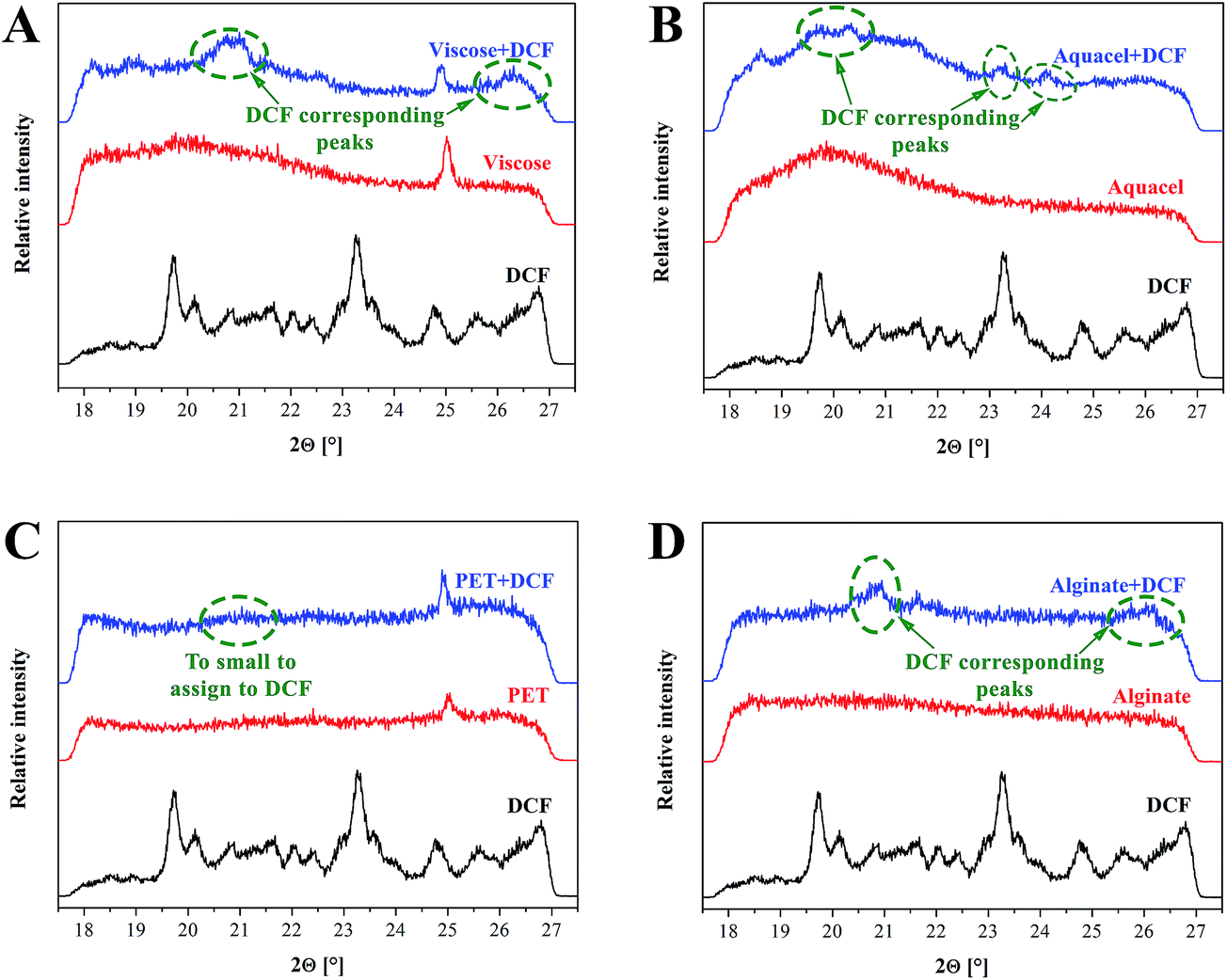 | ||
| Fig. 4 WAXS diffractograms of samples prior and after DCF incorporation, as well as the reference diffractograms of DCF. | ||
3.4. In vitro release studies
In this study the targeted application is wound healing, where the drug release rates have to efficiently follow the pharmacological specifics of different wound types. In vitro drug release testing is therefore a very important evaluation method in order to evaluate the prepared materials applicability in treatment of different wounds.Results from the in vitro release testing, performed using an automated Franz diffusion cell system, are depicted in Fig. 5. Only the first 360 minutes of release are presented, since this region exhibits the biggest differences between the used materials. The full release profiles are given in the ESI (Fig. S1†). The top figure (a) shows the mass of the released drug (g cm−2) for each DCF incorporated wound dressing material. The reason for using such units lies in the suitability of such representation for possible clinical application, where the dose can be easily calculated, based on the size of the dressing to be applied. The bottom figure (b) exhibits the percentage of the released drug as a function of time, which is important to immediately deduce the information about the release timeframe of the incorporated dose. Since, acutely released higher doses could possibly lead to unwanted side effects, such representation enables the planning of a safe and efficient treatment. In general, both types of representation of the in vitro release results are highly useful, and necessary to understand the release of the incorporated drug. Their combination renders planning of treatments for specific wound types possible. The total amounts of the incorporated DCF in the dressing after complete release (after 2880 minutes – 48 h) are presented in Fig. 6. Significant differences in release profiles can be observed in both representation types (Fig. 5a and b). This is also reflected in p-value (significant difference, see Table S1†), which was calculated using a stepwise regression analysis. A p-value of 0.05 is generally considered on the borderline of statistical significant difference. Thus, any p-value that is below 0.05 is usually regarded as statistically significant. Obviously, in our case, significant differences in release rates are observed for all wound dressing materials with incorporated DCF (Table S1†). Both alginate and Na-CMC samples, which are hydrophilic as proven by CA(H2O), showed a superior statistical significant differences (p < 0.001) compared to those of hydrophobic PET and viscose samples, where a p-value below 0.05 is obtained. The high p-values of viscose and PET samples indicates that their release profiles are certainly different as compared to that of other two materials such as alginate and Na-CMC, which can be clearly seen in Fig. 5a. Despite following the same DCF incorporation procedure, large differences were already observed in the amount of the incorporated drug per surface area of the material (Fig. 6). As mentioned above, these values are determined as the released amount after the drug concentration did not change anymore.
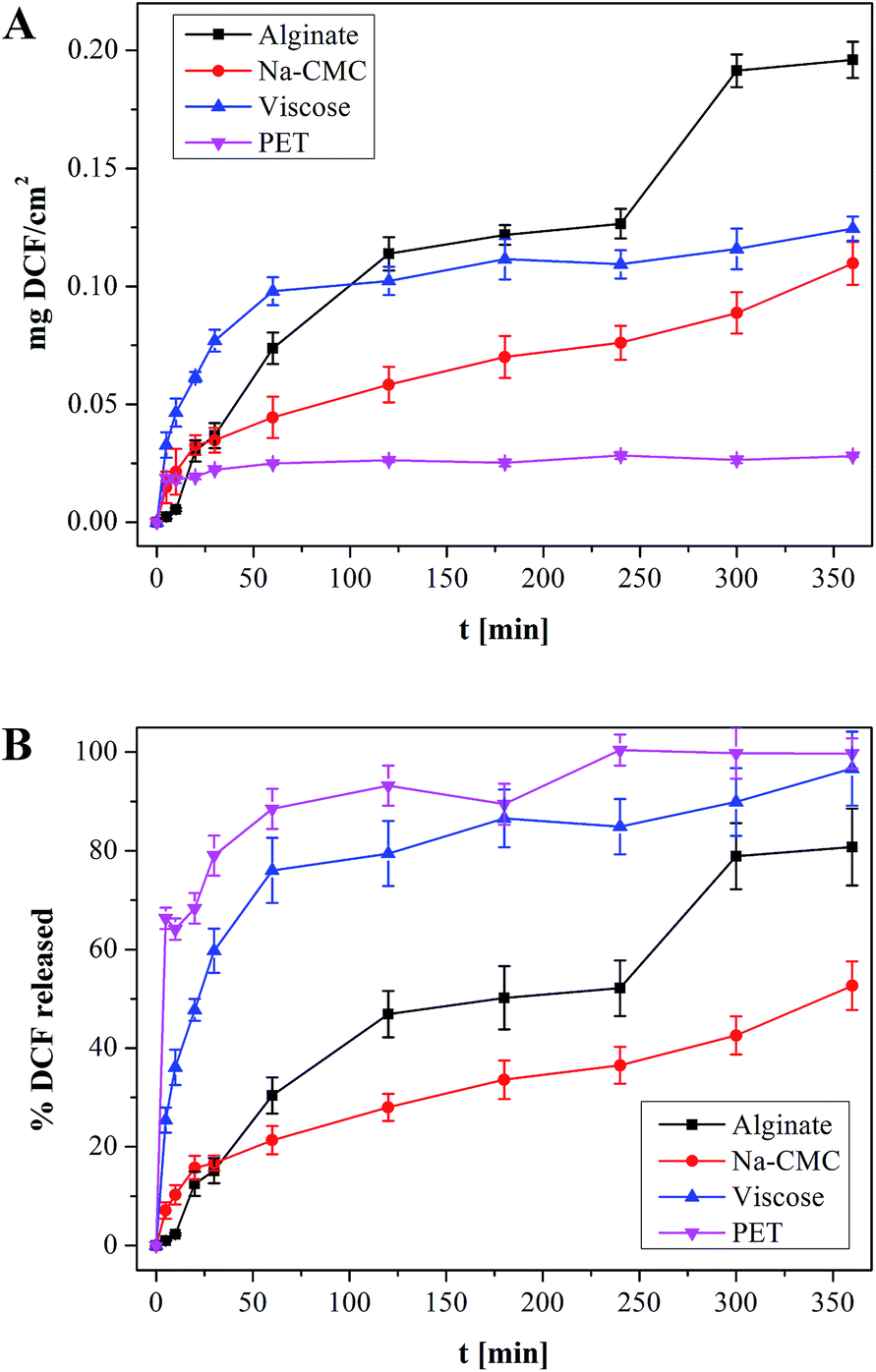 | ||
| Fig. 5 (a) amount of DCF released from a 1 × 1 cm squares model wound dressing and (b) percentage of released DCF, whereas the incorporated DCF amounts differ between the samples. Full release profiles are available as Fig. S1 in the ESI.† | ||
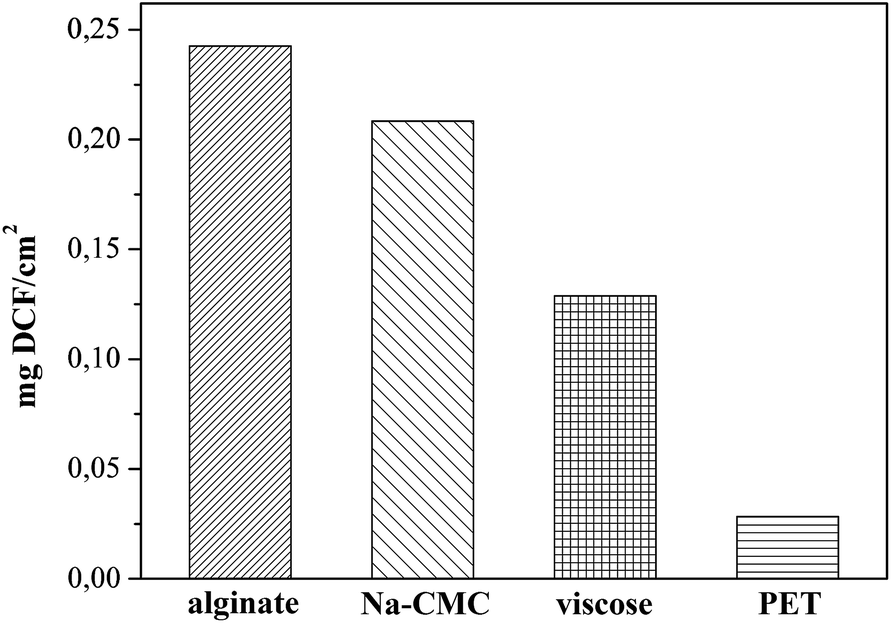 | ||
| Fig. 6 Total amounts of incorporated DCF in different wound dressing materials. | ||
Although the used drug in this study was not applied yet in a topical formulation clinically, the desired dose can be calculated based on the presently commercially available and clinically used DCF containing medicines for systemic use. The maximal dose in the latter is 75 mg per tablet. Since the bioavailability of the latter is in the range 30% locally, the desired dose in the wound would be approximately 23 mg. Considering the in vitro release results, an incorporation of DCF into a 10 × 10 cm squares alginate based dressing (alginate can incorporate, and hence release the maximal DCF amount), would lead to a maximum possible local concentration of around 20 mg. Considering the latter, we could claim that our formulations could be efficient, as well as safe. But only further clinical studies can confirm this assumption absolutely. As indicated in the Introduction section, the literature only sparsely reports the release from similar, whereas clinical studies are even more rare.12,13 There have been some reports about inclusion of ibuprofen, another NSAID into candidate wound dressing materials,34,36,52 not to forget about Biatain® IBU, the commercial, clinically used dressing with an incorporated NSAID.19,53 The latter served as a starting consideration point in our assumptions in regard of the dose.
While only a small drug amount (0.0282 mg cm−2) could be attached to PET, viscose (0.1289 mg cm−2), Na-CMC (0.2084 mg cm−2) and alginate (0.2426 mg cm−2) exhibited higher amounts of incorporated DCF (Fig. 6). Based on these results we are also able to judge the efficiency of the initial impregnation process, where the samples are able to soak the following percentages of DCF from solution, PET 2.8%, viscose 12.9%, Na-CMC 21%, and alginate 24.3%. Na-CMC and alginate can for example host almost an eight-fold larger amount of incorporated DCF compared to PET, which makes them more suitable for applications on chronic wounds, where the dressing change frequency is lower. On contrary, PET based dressings are probably not applicable in a single-layered form, since the incorporated amount of the drug does not cover the desired drug dose. Instead, considering Fig. 5b, PET could be very interesting for application on wounds with acute pain, where an immediate effect is necessary. The release profile shows a release of 60% of the incorporated drug in the first 5 minutes and reaches a plateau after 30 minutes. In the form of multi-layered dressings with PET as the bottom layer (in touch with skin/wound) could significantly add to the quality of patient treatment, since the pain would diminish immediately. Additionally, in such multi-layered dressings a PET based first layer would be interesting for treatment of wounds, where the dressing change frequency is high and is accompanied with pain, mainly caused by the removal of freshly epithelized skin or due to over-sensitization of the surrounding tissue as a consequence of inflammation.54
Interestingly, the viscose dressing shows a release profile similar to PET. However in the case of viscose 80% of the incorporated drug are released in the first 30 min, while the remaining 20% are released within 360 minutes. PET and viscose are very different chemically and structurally, as well as exhibit different wettabilities. These leads to a huge difference in drug uptake and importantly affects their possible usage in different applications. In addition, one must account for a rather complex structure of the material as a whole. Namely, viscose fibers are used in the form of a voluminous non-woven with plenty of space in-between individual fibers, enabling significant drug incorporation. This is not the case with the thin PET mesh, where only the actually exposed surface is capable to attach drug molecules. Its inert structure, as well as nonporous form with a low surface area, allow for attachment or incorporation of only small drug amounts (see Fig. 6). Nevertheless, PET is still an interesting material in wound care, since its inert surface prevents sticking to the wound. The most suitable application of a PET dressing is therefore as the initial layer in contact with the skin of multi-layered dressings. On the contrary, viscose in its non-woven form, allows on one hand the incorporation of a larger amount of DCF (in the range of 450% more, when compared to PET), and on the other, serves also a good absorbent for exudating wounds. The exudate can often lead to infections, significantly slowing wound healing. Viscose has therefore more options in regard of applicability. In can be used either as a lone dressing on highly exudating painful wounds or as the second layer of multi-layered dressings, serving as a drug reservoir in aid of the initial very fast release by the first PET layer.
Significantly higher doses of DCF could be incorporated into Na-CMC and alginate, also their release profiles and p-values (see Table S1, ESI†) are quite different from the other two materials (as discussed above), as well as differ also one from another. The latter is especially evident in the drug release rate differences. Na-CMC releases approximately 16% of DCF in 30 minutes, where the profile shows a turn and the release rate decreases. After the first 30 minutes, DCF is released at a constant rate. Making a linear fit of the curve from 30 minutes to the end, R2 value of 0.96 is obtained (not shown). These results suggest that incorporation of DCF into Na-CMC could assure a relatively constant DCF supply for at least 24 h. Such prolonged release characteristics of Na-CMC together with the highest capacity for binding and retaining water (see Fig. 1), is highly desired in case of chronic wound treatment, where pain is uninterruptedly present, the change of dressing is infrequent and an extensive exudation is present. A high amount of exudate is already known to limit the healing efficiency and therefore needs to be removed.41,55 Alginate on the other hand shows a 45% DCF release in the first two hours of release. After 240 minutes a sudden change in the release pattern is observed. In the next 60 minutes nearly 25% of DCF is released. An explanation for this sudden burst release can be that Na+ and Ca2+ in alginate structure are exchanged, leading to the breakdown of the base material mesh.56 This material degradation exposes an additional portion of the drug, which is then readily dissolved. The last 20% of the drug are then released in 24 h. Such materials and release performance could be interesting for wounds that are not moist enough on their own. Since alginate can hold a significant amount of water (see Fig. 1), the degradation of the material can lead to moisturizing of the wound bed. Release from alginate could be described as a combination of all the other observed profiles. Initial fast release during the first 2 h, followed by a diffusion controlled release with another burst of DCF release from 240 to 300 minutes. Using such host materials would be suitable for applications on wounds, where the pain caused by dressing change could be alleviated with a bolus dose and maintained through the following diffusion drug release, which would additionally reduce the injury induced pain sensation.
The results of the in vitro release are in accordance with other material properties, especially with the materials wettability. Viscose and especially PET exhibit high contact angles. And since DCF has a higher solubility in the used media, its fast release/dissolution in the media comes not as a surprise. Contrary, alginate with an intermediate hydrophilicity and a contact angle of 50°, which already incorporates a certain amount of water, releases the water soluble DCF with a smaller release rate. Na-CMC with its very high water retention value and an immeasurable contact angle, retains the drug even longer, since DCF has first to diffuse through the material and then only to the media. Both, alginate and Na-CMC form also gels (Na-CMC swells significantly), which also contributes to the smaller release rate than compared to the other two materials. Finally, all materials seem appropriate candidates for wound care, although for different wound types and PET only in combination with other materials. A schematic representation of the material–drug combinations in relation to the respective material in vitro release performance for wound care applications is shown in Fig. 7. To further verify the complete release of DCF, the samples collected after 24 h release are analyzed by ATR-IR and WAXS (see the ESI, Fig. S2 and S3†) measurements.
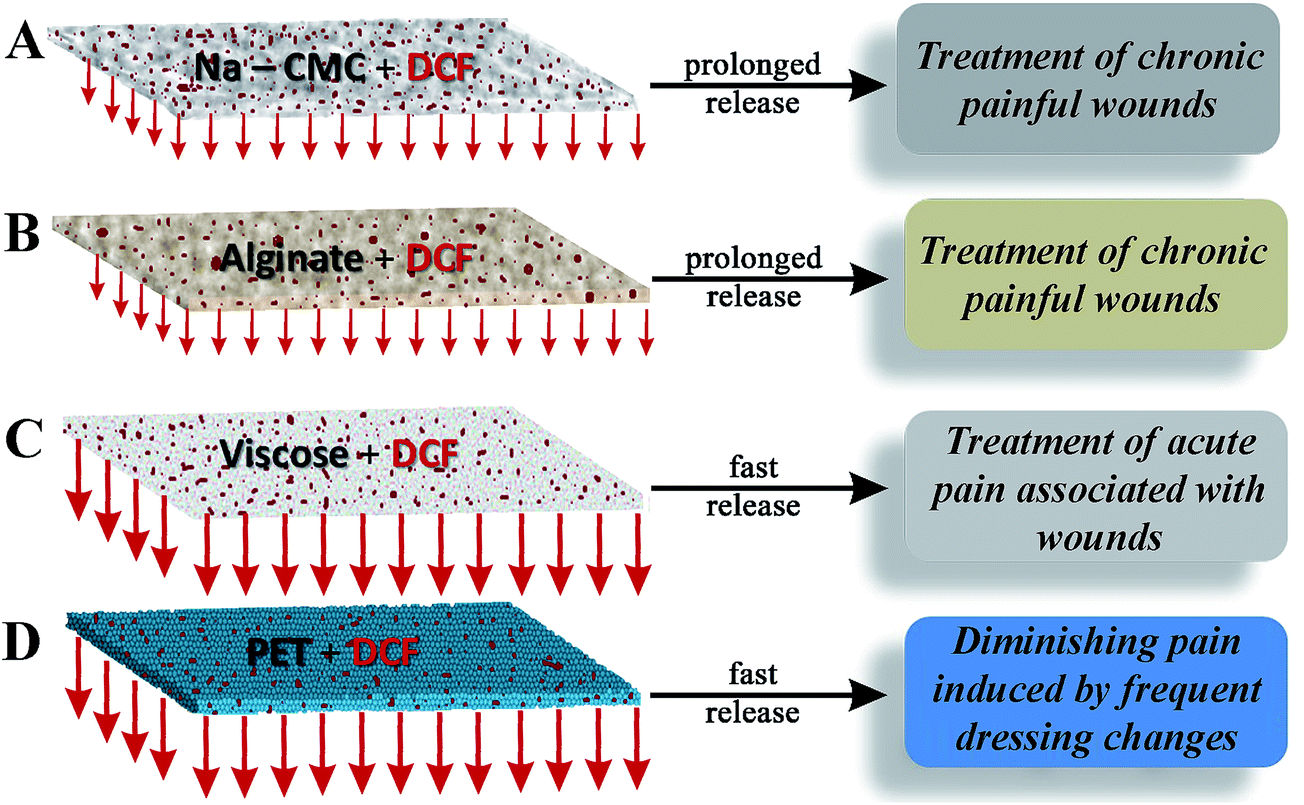 | ||
| Fig. 7 Schematic depiction of the material–drug combinations in vitro release performance and possible wound care applications. (a) Na-CMC with incorporated DCF suitable in treatment of chronic wounds, (b) alginate with incorporated DCF for treatment of chronic wounds with an even lower frequency of dressing change, (c) viscose with incorporated DCF for acute pain reduction and (d) PET with incorporated DCF as an initial layer in contact with the skin for wounds, requiring frequent dressing change. The drug DCF is depicted in red, while the materials are shown in different colors and morphologies, according to their macroscopic nature. Shorter arrows exhibit a prolonged release, while longer arrows correspond to a faster release. | ||
The ATR-IR and WAXS spectra showed no characteristic peaks that can be assigned to DCF in none of the used wound dressing materials collected after the in vitro release studies. The latter is a clear indication that all the incorporated drug was released.
3.5. SEM microscopy of sample surfaces prior and after in vitro release studies
Fig. 8 shows the SEM morphology of all materials prior and after drug incorporation, as well as after the in vitro release testing. SEM micrographs of pure drug particles are given in the ESI (Fig. S5†). All four chosen materials possess a fibrous form in their unloaded state (Fig. 8 – (U) left column). Some differences between materials are already evident after DCF incorporation (Fig. 8 – middle column). Na-CMC fibers seem to have been partially broken apart into block like parts and the presence of drug particles cannot be clearly observed. Another possibility is that this micrograph shows a broken part of the Na-CMC polymer film that formed after drug incorporation. All other materials retained their fibrous structure, while additional surface features can be observed that can be attributed to the loading of DCF. On the PET-based samples, only a small amount of drug particles could be observed on the surface, which is in agreement with the calculated (and measured) small amount of the incorporated drug. On the contrary, alginate and viscose samples exhibit clearly observable morphological changes on their respective surfaces. These corresponds well to the measured larger amounts of incorporated DCF during in vitro dissolution testing. While it seems that DCF is still in the form of crystals on the viscose fibers, a thin coating of DCF is visible on alginate samples. This is also in agreement with the results of in vitro release testing, where alginate clearly outperforms other materials in terms of the amount of the incorporated drug and release performance. Additional drug particles on the surface are smaller than crystals in the case of viscose (and PET), and as such, can improve the release rate also by increasing the effective surface area. | ||
Fig. 8 SEM micrographs of: left – the unloaded (U) materials, middle – alginate, Na-CMC, viscose and PET impregnated with DCF (+DCF), right – alginate, Na-CMC, viscose and PET after the release studies (after). The used magnification was 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×. 000×. | ||
Finally, SEM micrographs, taken after the in vitro drug release testing, are shown in Fig. 8 – right. While the morphology of viscose and PET based samples retained their initial fibrillar structure, there are significant differences evident for Na-CMC and alginate based samples. Alginate fibers are probably partially deformed through degradation as a result of cation (Na+ and Ca2+) exchange in its structure. This did not lead to severe material disintegration, but to an etching-like effect of the fiber surface as observed in Fig. 8 – right. The most notable changes after release are seen for Na-CMC, where the fibrous shape (and even the block-like structure, present after DCF incorporation) disappeared completely. Through exposure of Na-CMC to the dissolution media, Na-CMC swelled and formed a gel-like structure that rigorously differs from the initial fibered structure. All mentioned observations seem to correspond to the findings of other methods, especially with the results of in vitro drug release testing.
4. Conclusion
Recent economic trends, as well as ongoing rationalizations in health care, dictate the development of novel therapeutic approaches with lower overall costs without the sacrifice of patient safety. This can be achieved through optimized treatment efficiency and lowered hospitalization times. Wound care is no different from other health care sectors. We found that a combination of optimal materials and potent drugs could lead to great improvement in therapeutic efficiency of novel wound dressing materials, especially considering the different treatment approaches for specific wounds. Our results not only show that significant differences in the release profiles can be achieved by incorporating a NSAID, DCF into different materials, but also indicate the importance of a careful drug host material characterization in choosing the right material for the treatment of specific wounds.Acknowledgements
The authors acknowledge the financial support from the Ministry of Higher Education, Science and Technology of the Republic of Slovenia, as well as the financial contributions from the WoodWisdom-NET+ funded project AEROWOOD with the grant number 3330-14-500041 and mnt-era.net funded project WoundSens with the grant number 3211-12-00002.References
- T. Velnar, T. Bailey and V. Smrkolj, J. Int. Med. Res., 2009, 37, 1528–1542 CrossRef CAS PubMed.
- E. A. Gantwerker and D. B. Hom, Facial plastic surgery clinics of North America, 2011, 19, 441–453 CrossRef PubMed.
- L. G. Ovington, Clin. Dermatol., 2007, 25, 33–38 CrossRef PubMed.
- T. Pivec, Z. Peršin, M. Kolar, T. Maver, A. Dobaj, A. Vesel, U. Maver and K. Stana-Kleinschek, Text. Res. J., 2013, 84, 96–112 CrossRef PubMed.
- Z. Peršin, U. Maver, T. Pivec, T. Maver, A. Vesel, M. Mozetič and K. Stana-Kleinschek, Carbohydr. Polym., 2014, 100, 55–64 CrossRef PubMed.
- J. J. Elsner and M. Zilberman, Acta Biomater., 2009, 5, 2872–2883 CrossRef CAS PubMed.
- K. Vowden and P. Vowden, Surgery, 2014, 32, 462–467 Search PubMed.
- G. D. Mogoşanu and A. M. Grumezescu, Int. J. Pharm., 2014, 463, 127–136 CrossRef PubMed.
- N. Mayet, Y. E. Choonara, P. Kumar, L. K. Tomar, C. Tyagi, L. C. Du Toit and V. Pillay, J. Pharm. Sci., 2014, 103, 2211–2230 CrossRef CAS PubMed.
- M. Ignatova, I. Rashkov and N. Manolova, Expert Opin. Drug Delivery, 2013, 10, 469–483 CrossRef CAS PubMed.
- H. E. Thu, M. H. Zulfakar and S. F. Ng, Int. J. Pharm., 2012, 434, 375–383 CrossRef CAS PubMed.
- S. Kordestani, M. Shahrezaee, M. N. Tahmasebi, H. Hajimahmodi, D. Haji Ghasemali and M. S. Abyaneh, J. Wound Care, 2008, 17, 323–327 CrossRef CAS PubMed.
- G. Casey, Nurs. Stand., 2002, 17, 49–53 CrossRef , quiz 54, 56.
- J. S. Boateng, K. H. Matthews, H. N. E. Stevens and G. M. Eccleston, J. Pharm. Sci., 2008, 97, 2892–2923 CrossRef CAS PubMed.
- D. S. Katti, K. W. Robinson, F. K. Ko and C. T. Laurencin, J. Biomed. Mater. Res., Part B, 2004, 70, 286–296 CrossRef PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS.
- M. Kietzmann and M. Braun, Dtsch. Tierarztl. Wochenschr., 2006, 113, 331–334 CAS.
- M. D. Blanco, O. Garcia, C. Gomez, R. L. Sastre and J. M. Teijon, J. Pharm. Pharmacol., 2000, 52, 1319–1325 CrossRef CAS.
- B. Jørgensen, G. J. Friis and F. Gottrup, Wound Repair Regen., 2006, 14, 233–239 CrossRef PubMed.
- H. E. Thu, M. H. Zulfakar and S. F. Ng, Int. J. Pharm., 2012, 434, 375–383 CrossRef CAS PubMed.
- F. Gu, B. Amsden and R. Neufeld, J. Controlled Release, 2004, 96, 463–472 CrossRef CAS PubMed.
- N. Shimizu, D. Ishida, A. Yamamoto, M. Kuroyanagi and Y. Kuroyanagi, J. Biomater. Sci., Polym. Ed., 2014, 25, 1278–1291 CrossRef CAS PubMed.
- T. Muthukumar, K. Anbarasu, D. Prakash and T. P. Sastry, Colloids Surf., B, 2014, 121, 178–188 CrossRef CAS PubMed.
- P. Szweda, G. Gorczyca, R. Tylingo, J. Kurlenda, J. Kwiecinski and S. Milewski, J. Appl. Microbiol., 2014, 117, 634–642 CrossRef CAS PubMed.
- A. G. Richetta, C. Cantisani, V. W. Li, C. Mattozzi, L. Melis, F. de Gado, S. Giancristoforo, E. Silvestri and S. Calvieri, Recent Pat. Inflammation Allergy Drug Discovery, 2011, 5, 150–154 CrossRef CAS.
- J. R. Weiser and W. M. Saltzman, J. Controlled Release, 2014, 190, 664–673 CrossRef CAS PubMed.
- S. Moritz, C. Wiegand, F. Wesarg, N. Hessler, F. A. Müller, D. Kralisch, U.-C. Hipler and D. Fischer, Int. J. Pharm., 2014, 471, 45–55 CrossRef CAS PubMed.
- A. M. Abdelgawad, S. M. Hudson and O. J. Rojas, Carbohydr. Polym., 2014, 100, 166–178 CrossRef CAS PubMed.
- P. Price, K. Fogh, C. Glynn, D. L. Krasner, J. Osterbrink and R. G. Sibbald, Int. Wound J., 2007, 4(suppl. 1), 1–3 CrossRef PubMed.
- E. A. Gantwerker and D. B. Hom, Clin. Plast. Surg., 2012, 39, 85–97 CrossRef PubMed.
- S. Petrulyte, Dan. Med. Bull., 2008, 55, 72–77 CAS.
- K. Solowiej, V. Mason and D. Upton, J. Wound Care, 2010, 19, 110–115 CrossRef CAS PubMed.
- T. Maver, U. Maver, F. Mostegel, T. Griesser, S. Spirk, D. Smrke and K. Stana-Kleinschek, Cellulose, 2015, 22, 749–761 CrossRef CAS.
- K. Fogh, M. B. Andersen, M. Bischoff-Mikkelsen, R. Bause, M. Zutt, S. Schilling, J.-L. Schmutz, J. Borbujo, J. A. Jimenez, H. Cartier and B. Jørgensen, Wound Repair Regen., 2012, 20, 815–821 CrossRef PubMed.
- B. Steffansen and S. P. K. Herping, Int. J. Pharm., 2008, 364, 150–155 CrossRef CAS PubMed.
- L. Vinklarkova, R. Masteikova, D. Vetchy, P. Dolezel and J. Bernatoniene, BioMed Res. Int., 2015, 2015, 892671 Search PubMed.
- J. S. Boateng, H. V. Pawar and J. Tetteh, Int. J. Pharm., 2013, 441, 181–191 CrossRef CAS PubMed.
- H. V. Pawar, J. Tetteh and J. S. Boateng, Colloids Surf., B, 2013, 102, 102–110 CrossRef CAS PubMed.
- H. V. Pawar, J. S. Boateng, I. Ayensu and J. Tetteh, J. Pharm. Sci., 2014, 103, 1720–1733 CrossRef CAS PubMed.
- H. J. Jacobasch, K. Grundke, E. Mäder, K. H. Freitag and U. Panzer, J. Adhes. Sci. Technol., 1992, 6, 1381–1396 CrossRef CAS PubMed.
- A. D. Widgerow, Wound Repair Regen., 2011, 19, 287–291 CrossRef PubMed.
- C. Ramírez, I. Gallegos, M. Ihl and V. Bifani, J. Food Eng., 2012, 109, 424–429 CrossRef PubMed.
- R. Kargl, T. Mohan, M. Bračič, M. Kulterer, A. Doliška, K. Stana-Kleinschek and V. Ribitsch, Langmuir, 2012, 28, 11440–11447 CrossRef CAS PubMed.
- K. Stana-Kleinschek, V. Ribitsch, T. Kreze, M. Sfiligoj-Smole and Z. Persin, Lenzinger Ber., 2003, 82, 83–95 Search PubMed.
- W. E. Morton and J. W. S. Hearle, Physical Properties of Textile Fibres, 4th edn, 2008 Search PubMed.
- L. Fan, Y. Du, R. Huang, Q. Wang, X. Wang and L. Zhang, J. Appl. Polym. Sci., 2005, 96, 1625–1629 CrossRef CAS PubMed.
- D. W. van Krevelen and K. Te Nijenhuis, Properties of Polymers, 2009 Search PubMed.
- I. Bravo-Osuna, C. Ferrero and M. R. Jiménez-Castellanos, Eur. J. Pharm. Biopharm., 2008, 69, 285–293 CrossRef CAS PubMed.
- F. K. Field and M. D. Kerstein, Am. J. Surg., 1994, 167, 2S–6S CrossRef CAS.
- A. Miyake, J. Polym. Sci., 1959, 38, 479–495 CrossRef CAS PubMed.
- C. F. Fan and T. Cagin, J. Chem. Phys., 1995, 103, 9053–9061 CrossRef CAS PubMed.
- M. Romanelli, V. Dini, R. Polignano, P. Bonadeo and G. Maggio, J. Dermatol. Treat., 2009, 20, 19–26 CrossRef CAS PubMed.
- F. Gottrup, B. Jørgensen, T. Karlsmark, R. G. Sibbald, R. Rimdeika, K. Harding, P. Price, V. Venning, P. Vowden, M. Jünger, S. Wortmann, R. Sulcaite, G. Vilkevicius, T.-L. Ahokas, K. Ettler and M. Arenbergerova, Int. Wound J., 2007, 4, 24–34 CrossRef PubMed.
- G. Wozniak, P. Mauckner, L. Steinstrasser and J. Dissemond, Gefässchirurgie, 2011, 16, 281–290 CrossRef.
- D. Ulrich, R. Smeets, F. Unglaub, M. Wöltje and N. Pallua, Journal of Wound Ostomy & Continence Nursing, 2011, 38, 522–528, DOI:10.1097/won.1090b1013e31822ad31290.
- C. H. Goh, P. W. S. Heng and L. W. Chan, Carbohydr. Polym., 2012, 87, 1796–1802 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra11972c |
| This journal is © The Royal Society of Chemistry 2015 |