
Catalytic hydrogenolysis of glycerol to propanediols: a review
Yanli Wang
a,
Jinxia Zhou
*b and
Xinwen Guo
*a
aState Key Laboratory of Fine Chemicals, PSU-DUT Joint Center for Energy Research, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, PR China. E-mail: guoxw@dlut.edu.cn; zhoujxmail@163.com; Fax: +86 0411 84986134, +86 0411 87402449; Tel: +86 0411 84986133, +86 0411 87403214
bCollege of Environmental and Chemical Engineering, Dalian University, Dalian, 116012, PR China
First published on 14th August 2015
Abstract
The catalytic hydrogenolysis of readily available glycerol to 1,2-propanediol (1,2-PD) and 1,3-propanediol (1,3-PD), which provides a new promising synthesis route to produce propanediols, has been extensively studied in the past decades. This study summarizes the most significant reports regarding glycerol hydrogenolysis into propanediols. Three reaction routes, including those working towards 1,2-PD production and the recently proposed one leading to 1,3-PD production, have been summarized. The catalysts used for this reaction have been classified into two categories according to the type of metal components: the transition metal catalysts taking Cu, Ni, and Co as representative metal components and the noble metal catalysts containing Ru, Pt, Ir, and Ag. Some inexpensive transition-metal catalysts exhibit high 1,2-PD selectivity and yield under mild reaction conditions, whereas several noble metal catalysts are promising in synthesizing the more valuable 1,3-PD. Efficient preparation methods and precise modulation techniques have been systematically developed to synthesize functionalized catalysts on the basis of the metal species in combination with acidic or basic compounds. Other technological aspects, such as hydrogen sources, reaction solvents, reactor types and feeding processes, are also summarized in this study. The focus of this review is on summarizing the preparation methods and the performance of various catalysts in glycerol hydrogenolysis.
 Yanli Wang | Yanli Wang obtained his B.S. and M.S. from the Yanching Institute of Technology and the Beijing University of Chemical Technology in 2007 and 2011, respectively. Since 2014, he has been pursuing his Ph.D. at the Dalian University of Technology under the guidance of Prof. Xinwen Guo. His current research topic is the synthesis of Ti-containing molecular sieves for catalyzing the epoxidation of propylene. |
 Jinxia Zhou | Jinxia Zhou obtained her Ph.D. in Industrial Catalysis in 2007 from the Dalian University of Technology under the guidance of Prof. Xinwen Guo. Then, she moved to the Dalian University and engaged in teaching and research work. From 2012–2014, she joined the research group of Prof. Z. Conrad Zhang at the Dalian Institute of Chemical Physics, Chinese Academy of Sciences for two years of postdoctoral research. Her current research interests are developing platform chemicals and biofuels from biomass. |
 Xinwen Guo | Xinwen Guo received his Ph.D. in Industrial Catalysis in 1994 from the Dalian University of Technology. He became a lecturer (1994–1996) and an associate professor (1996–2001) of the major Industrial Catalysis in the Dalian University of Technology. In 2001 he became a professor, working in the Faculty of Chemical, Environmental and Biological Science and Technology, Dalian University of Technology. His current research interests are the synthesis of hierarchical materials and catalytic conversions of methanol and CO2 as well as epoxidation of olefins. |
1. Introduction
The anticipated depletion of petrochemical resources and the increasing awareness of environmental problems motivate the use of sustainable resources. Biomass, which is a biological material derived from living organisms, can serve as a partial replacement feedstock for petroleum-derived chemicals and liquid fuels.1–4 Glycerol is a biomass resource and a natural chemical, which serves as a linker molecule in most animal and plant oils and fats. It can be produced from steam splitting or saponification processes such as the traditional soap manufacturing (Scheme 1, Route A). It is also now available as a major byproduct of biodiesel production processes (Scheme 1, Route B), in which glycerol amounts to about 10 wt% of the total biodiesel produced. Moreover, glycerol production through biological fermentation processes based on inexpensive and abundant materials and cellulolytic derivatives is booming quickly and will become another useful route for the production of glycerol.5–8 With the escalating production and application of biomass, glycerol has become more readily available as a renewable resource.9,10 Therefore, great interest has been taken in the development of novel technologies to convert glycerol to value-added chemicals like propanediols. | ||
| Scheme 1 Generation of glycerol by saponification and biodiesel processes. | ||
Propanediols are important commodity chemicals. 1,2-PD11 has a valuable use as a less toxic alternative to other chemicals in paint, liquid detergent, cosmetics, food and tobacco and can be used as an antifreeze coolant and a de-icing reagent. It is also extensively used as feedstock in the preparation of polyester resins for film, in fiber manufacture and in the pharmaceutical industry. 1,3-PD9,12 is mainly used as a monomer in the manufacture of polymers such as polytrimethylene terephthalate (PTT, a biodegradable polyester that has great potential for use in carpet and textile manufacturing), polyethers and polyurethanes. Even though various approaches, such as fermentation, hydroformylation–hydrogenation and hydration methods,13–15 are applied to manufacture propanediols by some companies, propanediols are most commonly produced from propylene via a process that involves selective propylene oxidation to propylene oxide and subsequent hydrolysis. The supply of propylene derived from crude oil has been restricted, motivating urgent development of a more economical production from renewable alternative feeds.
Glycerol can be converted into propanediols via catalytic hydrogenolysis, which provides a new promising synthesis route. The reactions and processes for catalytic hydrogenolysis of glycerol to propanediols have been extensively studied in the past decades. A systematic summary of the existing experiments and achievements provides valuable guidance for further study. This review summarizes the most significant reports regarding the production of propanediols via glycerol hydrogenolysis, including the proposed mechanisms, developed catalysts and some special process conditions.
2. Mechanisms
As glycerol has one more –OH groups than propanediols, hydrogenolysis of glycerol to propanediols consists of the removal of one hydroxyl group and the addition of one hydrogen or, equivalently, the removal of one H2O and the addition of one H2 molecule.12 The removal of the side –OH group of glycerol forms 1,2-PD, whereas dissociation of the middle –OH group generates 1,3-PD. The competing reactions in glycerol hydrogenolysis include the breaking of one C–C bond to form ethylene glycol (EG). Over-hydrogenolysis of C–C and C–O bonds in glycerol or in its derived products will further generate monobasic alcohols and alkanes.14 Scheme 2 summarizes the possible products of the glycerol hydrogenolysis reaction, including the main products, 1,2-PD and 1,3-PD, as well as many byproducts such as EG, propanols, ethanol, methanol and methane.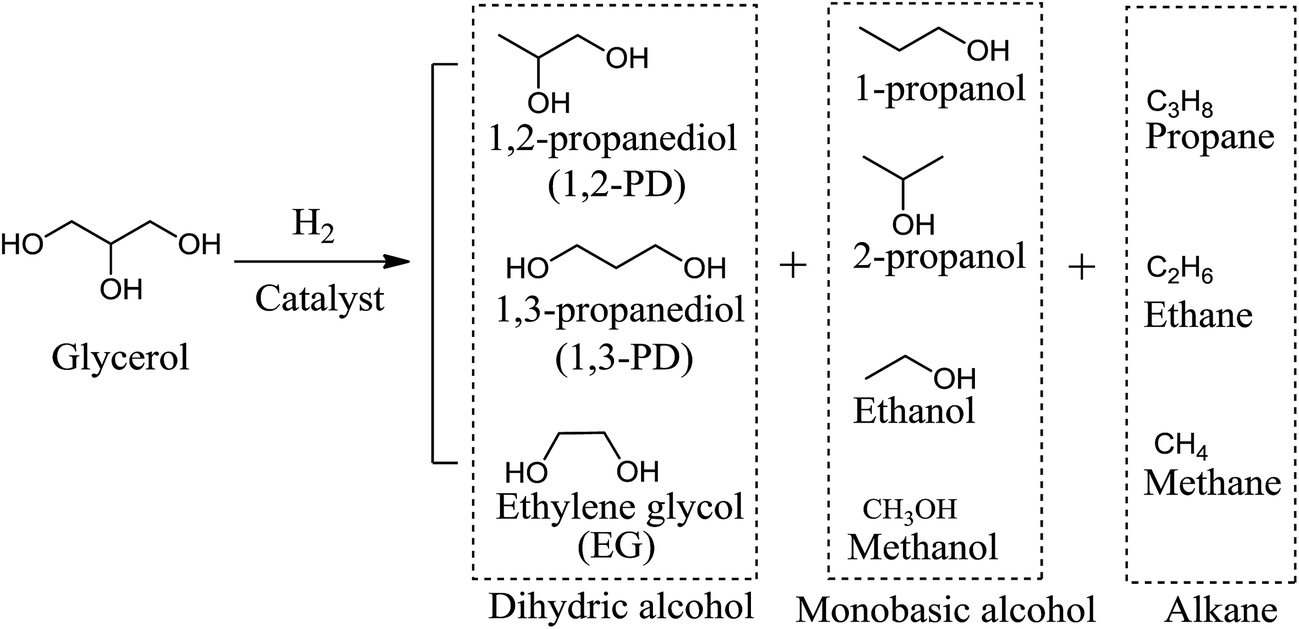 | ||
| Scheme 2 Potential products of glycerol hydrogenolysis grouped by the extent of hydrogenation. | ||
Many mechanisms corresponding to glycerol hydrogenolysis have been proposed, and the operating mechanism in any given process may depend on the properties of the reaction systems such as acidic or basic catalytic properties or metal catalytic properties. There are three typical reaction mechanisms (Scheme 3) that have been generally accepted, namely, dehydration–hydrogenation, dehydrogenation–dehydration–hydrogenation and a recently proposed direct-hydrogenolysis mechanism.
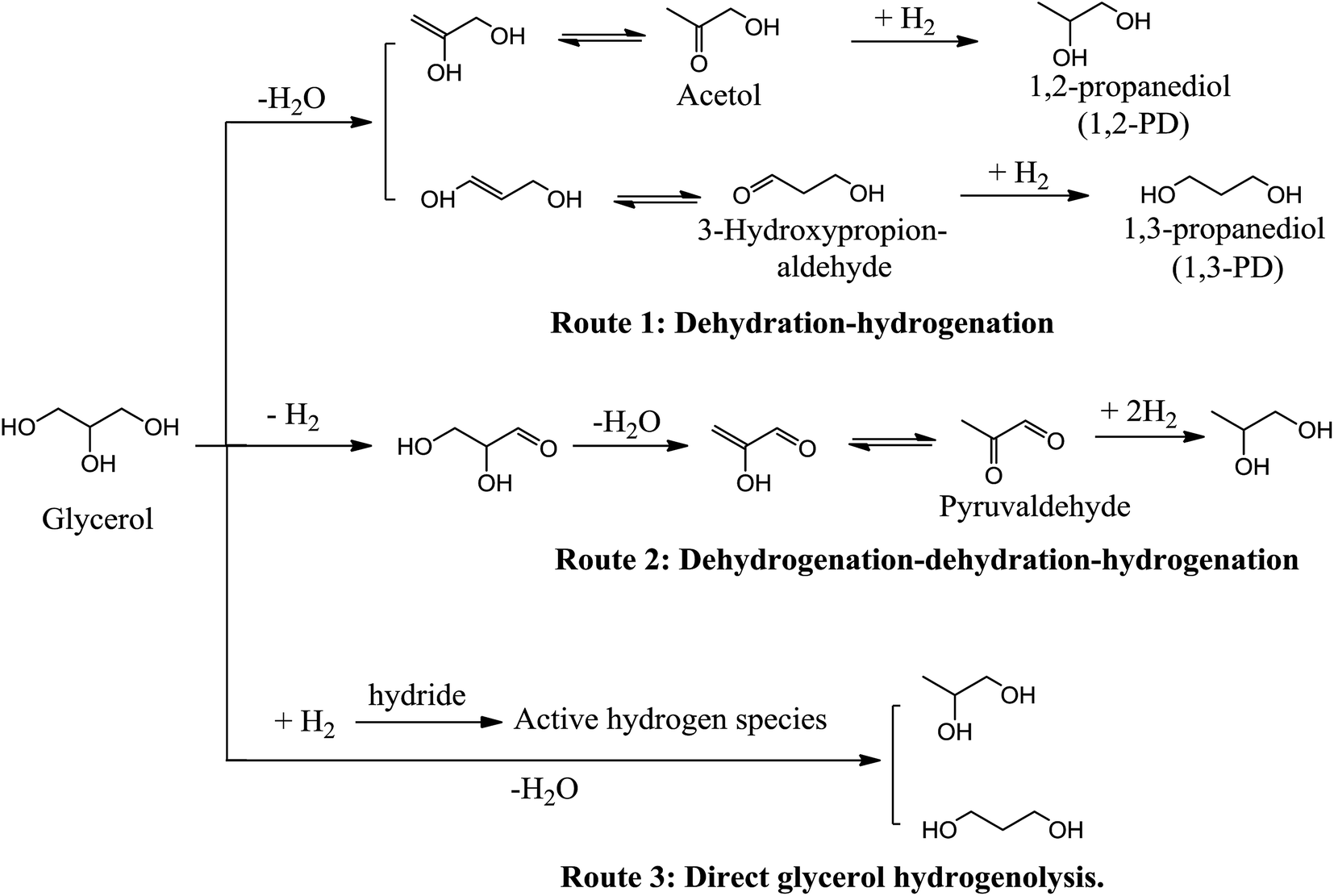 | ||
| Scheme 3 Proposed mechanisms of glycerol hydrogenolysis to produce propanediols. | ||
2.1. Dehydration–hydrogenation route
In the dehydration–hydrogenation mechanism, glycerol is initially dehydrated through an acid-catalyzed reaction to form an intermediate and then the intermediate is subsequently hydrogenated to generate the final product. When a proton attacks the –OH linked to the terminal carbon of glycerol, the dehydrated intermediate is acetol, which is further hydrogenated to 1,2-PD (Scheme 3, Route 1). The acetol intermediate has been collected and confirmed from glycerol dehydration by a reactive distillation process using copper chromite catalysts and then hydrogenated to form 1,2-PD using the same catalyst.16 Accordingly, 1,3-PD can be formed from the dehydration of the middle –OH of glycerol to form 3-hydroxypropionaldehyde, followed by subsequent hydrogenation. Tyrlik et al.17 and Miyazawa et al.18 have proposed that –OH on a Ru/C catalyst attacks the H linked to the terminal carbon of glycerol, which induces glycerol dehydration to form 3-hydroxypropionaldehyde (Scheme 3, Route 1). Based on this proposal, 1,3-PD can be generated by hydrogenation of 3-hydroxypropionaldehyde formed by dehydration of glycerol. Although both 1,2-PD and 1,3-PD can be theoretically generated in the dehydration–hydrogenation route, propanols are always produced as major byproducts and are the main cause of reduced propanediol selectivity. 1-Propanol is generated by the hydrogenation of acrolein formed by the acid-catalyzed dehydration of glycerol and over hydrogenolysis of 1,2-PD and 1,3-PD, whereas 2-propanol forms through the over-hydrogenolysis of the –OH linked to the terminal carbon of glycerol. A common feature of these studies is the low probability of forming 1,3-PD, which can be attributed to the much higher stability of the intermediate acetol compared with 3-hydroxypropionaldehyde.192.2. Dehydrogenation–dehydration–hydrogenation route
The dehydrogenation–dehydration–hydrogenation mechanism (Scheme 3, Route 2) usually takes place in neutral water and alkaline conditions. The reaction route follows three steps containing dehydrogenation, dehydration and hydrogenation. Montassier et al.20,21 adopted a series of ruthenium catalysts for glycerol hydrogenolysis and suggested that glycerol is initially dehydrogenated to form glyceraldehyde on the metal sites of the catalysts, followed by dehydration of the glyceraldehyde intermediate on the base sites to form 2-hydroxyacrylaldehyde, which hydrogenates sequentially on the metal sites to form acetol and propylene glycol. Apart from the scission of one C–O bond leading to propanediol formation, C–C bond cleavage to form ethylene glycol (EG) and C1 byproducts cannot usually be neglected when glycerol hydrogenolysis is conducted under alkaline conditions.22 As shown in Scheme 4, glyceraldehyde derived from glycerol dehydrogenation can undergo decarbonylation on the noble metal surfaces to form ethylene glycol and CO or undergo retro-aldolization reactions on the basic sites to form the 2-hydroxyacetaldehyde and formaldehyde intermediates, which are then hydrogenated over the metals to yield EG and methanol, respectively. Hydrogenolysis of methanol and hydrogenation of CO on the metals result in the formation of methane. Moreover, CO may convert to CO2 via a water gas shift reaction. | ||
| Scheme 4 Mechanism for the formation of EG and C1 byproducts. | ||
2.3. Direct-hydrogenolysis route
A direct-hydrogenolysis mechanism was recently proposed by Tomishige et al.23–25 The authors suggested that this mechanism applies to glycerol hydrogenolysis on Ir–ReOx/SiO2 catalysts. Initially, glycerol is adsorbed on the interface between the Ir metal surface and the ReOx cluster to form two terminal alkoxides: 2,3-dihydroxypropoxide (Scheme 5a) and 1,3-dihydroxyisopropoxide (Scheme 5b). Then, hydride activated on the Ir metal attacks the 2-position of 2,3-dihydroxypropoxide and the 3-position of 2,3-dihydroxypropoxide to break the C–O bond. Finally, the hydrolysis of the reduced alkoxide releases the product. | ||
| Scheme 5 Model structures of the transition states of the hydride attack to the adsorbed substrate in glycerol hydrogenolysis.25 | ||
The proposed structure of 2,3-dihydroxypropoxide is more stable as the formation of 1,3-dihydroxyisopropoxide forms an unstable 7-membered ring. The hydride attacks the 2-position of 2,3-dihydroxypropoxide to give 1,3-PD, which is formed through the more stable 6-membered-ring transition state and 3-hydroxypropoxide (Scheme 5a). Subsequently, the hydride attack on the 3-position of 2,3-dihydroxypropoxide leads to 1,2-PD via an unstable 7-membered-ring transition state and 2-hydroxypropoxide (Scheme 5b). The transition state in the hydrogenolysis of 1,3-PD (Scheme 5c) is similar to 1,3-dihydroxyisopropoxide, which is unstable and not easily formed. The transition state occurring during the formation of 1,2-PD (Scheme 5d) is relatively stable. Therefore, the lower selectivity to 1,2-PD and its over-hydrogenolysis products compared to that of 1,3-PD in this system can be explained by the high stability of the 6-membered-ring transition state.
3. Transition metal catalysts
Catalysts for the hydrogenolysis of glycerol have two catalytic functions, namely, acidic or basic functionality for removal of an –OH group and oxidation–reduction functionality for addition of hydrogen. Traditionally, the metal components of the catalysts play roles in activating hydrogen and the metal oxides or acidic or basic supports are used to provide the acid–base function. The catalysts used for this reaction are typically classified into two categories based on the type of metal used: transition metal catalysts and noble metal catalysts. Transition metal catalysts have been intensively investigated in the hydrogenolysis of glycerol, and Cu, Ni and Co are the most frequently utilized metal components.3.1 Cu-based catalysts
| Catalyst | Preparation method | T (K)/P (MPa,H2)/time (h) | Water (g)/glycerol (g)/catalysta (%) | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|
| 1,2-PD | ||||||
| a With respect to glycerol mass unless particularly stated.b 2-Propanol as the solvent.c Isopropanol as the solvent. | ||||||
| Cu/ZrO2 | Co-precipitation | 473/4.0/8 | 15/10/6 | 11.4 | 92.4 | 26 |
| BaCuO/CuCr2O4 | Co-precipitation | 493/5.2/5 | 20 wt%/0.01b g mL−1 | 34.0 | 85.0 | 28 |
| CuO/CuCr2O4 | Co-precipitation | 493/6.0/12 | 5/45/2.22 | 78.3 | 75.9 | 29 |
| PdCuO/CuCr2O4 | Co-precipitation | 493/4.0/18 | 5/45/2.22 | 93.9 | 100 | 29 |
| CuO/SiO2 | Precipitation–gel | 473/9.0/12 | 16/64/6.25 | 73.4 | 94.3 | 30 |
| CuO/SiO2 | Precipitation–gel | 453/9.0/12 | 8/32/6.25 | 32.7 | 98.7 | 31 |
| CuO/SiO2 | Heterogeneous deposition–precipitation | 453/9.0/12 | 16/64/6.25 | 35.0 | 93.7 | 32 |
| Cu/Al2O3 | Co-precipitation | 493/7.0/5 | 83.69/20.52/3.82 | 38.0 | 91.0 | 33 |
| Cu/Al2O3 | Co-precipitation | 493/5.2/5 | 83.69/20.52/4.78 | 63.0 | 88.0 | 34 |
| Cu/Al2O3 | Co-precipitation | 473/4.0/24 | 4.86/19.43/5 | 75.7 | 95.8 | 35 |
| Cu/MgO | Co-precipitation | 473/4.0/8 | 41.85/10.46/5.74 | 49.3 | 92.3 | 36 |
| CuO/MgO | Co-precipitation | 453/3.0/20 | 2.40/7.19/13.91 | 72.0 | 97.6 | 37 |
| Cu/ZnO | Co-precipitation | 473/2.0/16 | 83.69/20.52/5.74 | 37.0 | 92.0 | 38 |
| Cu/fly ash | Precipitation | 493/5.2/5 | 83.7/20.5/4.8c | 57.0 | 94.0 | 39 |
| CuZnMgAl2O2 | Co-precipitation | 453/2.0/10 | 2/6/0.17 | 78.2 | 99.3 | 40 |
| PdCuMgAl2O3 | Co-precipitation | 453/2.0/10 | 2/6/0.17 | 95.0 | 96.1 | 41 |
Copper chromite,27 a type of co-precipitated CuCr catalyst, has been used as a catalyst for the conversion of glycerol to 1,2-PD. The reduced Cu metal component activates hydrogen and the chromium oxides are used to provide the acidic functionality. Although copper chromite has significant catalytic activity, the selectivity to 1,2-PD is not over 90%. CuO–Ba/CuCr2O4 catalysts prepared using an ammonium chromate and Cu(NO3)2 solution and modified by Ba show better performances, which are attributed to the enhanced acidity and stabilization of Cu0 species promoted by BaCrO4.28 Another enhancement in the performance of a CuO/CuCr2O4 catalyst is achieved by adding 0.5 wt% Pd for the conversion of glycerol to 1,2-PD.29 Pd was observed to enhance the amounts of surface-exposed Cu0 species and occluded hydrogen species. As a result, the reaction catalyzed by the Pd–CuCr catalyst can be performed at a relatively low hydrogen pressure with high catalytic activity. The Pd0.5–CuO/CuCr2O4 catalyst (containing 0.5 wt% of Pd) showed a total yield of 93.9% with a selectivity approaching 100% for 1,2-PD at a hydrogen pressure of 4 MPa.
Cu/SiO2 catalysts with high Cu dispersion were synthesized by a precipitation method and a good 1,2-PD yield was obtained.30–32 The catalyst was prepared by adding suitable quantities of NaOH and colloidal aqueous silica into a Cu(NO3)2 solution. TEM and XRD results illustrate that the sample, after calcination at 723 K, presents highly dispersed CuO and Cu–Si spinel with an average particle diameter of 6 nm, which gives an excellent performance with a 1,2-PD yield of 69.2%.30 However, the residual sodium on the catalyst has a detrimental effect on surface area, Cu dispersion, reducibility of Cu2+ and affinity to reactant molecules. This eventually lowers the 1,2-PD yield despite its positive impact on the stability of Cu/SiO2,31 indicating that the effects of the precipitant are crucial to the chemical–physical properties of the catalyst. They further investigated the effect of the precipitation manner on the properties of the catalyst.32 TEM images suggest that the catalyst prepared by homogeneous precipitation (adding colloidal aqueous silica into a cuprammonia complex) exhibits smaller particle sizes (4.2–4.7 nm) and higher Cu dispersion compared with the heterogeneous method (6.1–7.0 nm), in which dried silica gel powder was added into the solution of Cu(NO3)2 and then precipitated by adding an aqueous solution of NaOH. However, homogeneous precipitated catalysts exhibit inferior catalytic activity of only 25.0% conversion, which is attributed to the lower content of active Cu0 sites in the reduced catalyst.
A series of Cu/Al2O3 catalysts were prepared by adding aqueous potassium carbonate into an aqueous solution of Cu(NO3)2 and Al(NO3)3. Catalysts with an impressive 1,2-PD selectivity of around 90% were obtained by calcinating the precipitate in air, followed by subsequent pre-reduction in H2.33–35 Mane et al. pointed out that Cu+ generated after pre-reduction can stabilize the particle size in a narrow range and prevent sintering of Cu0 under the given reaction conditions.33 They investigated the effect of precipitating agents and preparation methods on physico-chemical properties of Cu/Al2O3 catalysts.34 Catalysts prepared by a co-precipitation method with Na2CO3 as the precipitating agent exhibit stronger acidity, lower aggregation and better reducibility compared with the catalysts prepared by alkali fusion followed by precipitation and direct solid state fusion methods. Instead of K+, Na+ increases the acidity due to the alkali metal trend, and the corresponding catalyst also presents smaller metallic Cu particles, which resulted in improving the 1,2-PD yield to 55.4% at a lower pressure. In addition, Milchert et al.35 believe that an excessive reaction temperature, time, catalyst concentration, glycerol concentration and stirring speed have detrimental effects on 1,2-PD selectivity due to the occurrence of secondary reactions, such as C–C bond cleavage, over-hydrogenolysis and even coke deposition, implying that proper operation conditions are necessary for high catalytic performance.
A Cu–MgO catalyst36,37 achieved considerable 1,2-PD selectivity by the route presented in Scheme 3, Route 2. However, an inferior performance was achieved when the Cu loading was higher than 20 wt%. Since strong basicity and higher Cu dispersion can efficiently activate glycerol and H2, CuO clusters in high Cu-content catalysts not only cover many of the MgO basic sites but also lower the Cu dispersion, resulting in a lower activity of the catalyst.36 The particle size of MgO also had a significant influence on the catalytic activity.37 Superior glycerol conversion is achieved on the sample with smaller CuO and MgO particles (4.0 nm and 25.6 nm, respectively), implying that good dispersion of metal components is very important. Similar to the Cu–MgO catalyst, a Cu–ZnO catalyst with 50 wt% Cu content showed the smallest Cu and ZnO particle sizes of 33.4 nm and 30.9 nm, respectively, and thus exhibited the best catalytic performance compared with other samples in the same category.38 Nevertheless, Cu particle size in Cu–ZnO is still larger than that in Cu–MgO catalysts and thus the performance is inferior in comparison with Cu–MgO catalysts, which is related to the intrinsic property of the support.36–38 This implies that methods of support selection and modification are necessary for metal dispersion enhancement. When using ZnO, MgO and Al2O3 together, the Cu0.4Zn0.6Mg5.0Al2O8.6 catalyst exhibits a better catalytic performance compared with mono-solid acid or base catalysts.40 A multi-component catalyst, PdxCu0.4Mg6−xAl2O9−x, synthesized by the simultaneous adoption of multiple metals and metal oxides, has achieved a superior catalytic performance.41 Pd and Cu were highly dispersed in the reduced bimetallic Pd–Cu/solid-base catalyst and a small amount of Pd improved the reduction of copper oxides. On Pd0.04Cu0.4/Mg5.56Al2O8.56, the conversion of glycerol and the selectivity to 1,2-PD reached 95.0% and 96.1%, respectively, at 2.0 MPa H2, 453 K, 10 h. It was concluded that H2-spillover from Pd to Cu increased the activity of PdxCu0.4/Mg5.6−xAl2O8.6−x in the hydrogenolysis of glycerol.41
| Catalyst | Preparation method | T (K)/P (MPa,H2)/time (h) | Water (g)/glycerol (g)/catalysta (%) | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|
| 1,2-PD | ||||||
a With respect to glycerol mass unless particularly stated.b Hexagonal mesoporous silica.c N-Butanol glycerol solution.d WHSV, H2/glycerol = 123![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 4 g catalyst, 2-propanol served as solvent.e Molar ratio of (Cu + Ag)/glycerol. :1, 4 g catalyst, 2-propanol served as solvent.e Molar ratio of (Cu + Ag)/glycerol. |
||||||
| Cu/HMS silicateb | Impregnation | 513/8.0/5 | 0/153/0.6 | 43.0 | 91.1 | 42 |
| Cu/SiO2 | Impregnation | 513/8.0/5 | 58.42/61.58/0.6c | 51.9 | 96.6 | 43 |
| Cu-delaminated hectorites | Impregnation | 473/4.0/8 | 20.75/13.83/7.23 | 61.4 | 93.0 | 44 |
| Cu/B2O3/SiO2 | Impregnation | 473/5.0/- | 10 wt%/0.075d h−1 | 100 | 98.0 | 45 |
| Cu/Al2O3 | Impregnation | 493/3.6/10 | 32.5/32.5/2(Cu) | 49.6 | 96.8 | 46 |
| CuAg/Al2O3 | Impregnation | 473/3.6/10 | 22.5/22.5/3e | 27.0 | 96.0 | 48 |
Cu–Si catalysts with various silica types, such as hexagonal mesoporous silica, commercial silica gel, SBA-15, and delaminated hectorite, have been applied in glycerol hydrogenolysis to 1,2-PD.42–45 The type of silica mainly affects the Cu dispersion, the active copper metal area, and the catalytic activity and stability of the catalysts. The correlation of TOF results with Cu crystallites indicates that glycerol hydrogenolysis over Cu catalysts is a structure-sensitive reaction. Deactivation of Cu–Si catalysts is commonly attributed to Cu aggregation or coverage of the catalytic surface by adsorbed species and coke, rather than to Cu leaching. Collapse of the mesoporous network is another factor causing Cu/HMS silicate deactivation.42 Vasiliadou et al.43 suggested that a Cu/SBA catalyst calcined in air exhibits a larger active Cu surface area, smaller Cu particle size and better reusability compared with the one calcined in a mixture of nitric oxide and nitrogen, indicating that the calcination atmosphere plays a significant role in both the activity and stability of Cu–Si catalysts. B2O3 (3 wt%), a non-metallic oxide, was added to improve the stability of a Cu/SiO2 catalyst and it exhibited an excellent catalytic performance during a 150 hour experimental run. The close linear relationship between 1,2-PD yield and copper surface area as well as the strong correlation between TOF and Cu particle size revealed that a higher yield was achieved with a higher Cu dispersion. The Cu dispersion could be adjusted by varying the B content. Moreover, interaction between copper species and B2O3 and the formation of cupric phyllosilicate inhibited the sintering of metallic Cu nanoparticles during the pretreatment and reactions. As a result, Cu–B/SiO2 catalysts have a longer lifespan than Cu/SiO2 catalysts (30 h), suggesting that the non-metallic oxide can also be considered as a dopant to enhance catalyst stability.44,45
Guo et al.46 prepared a supported Cu catalyst using an impregnation method to improve the efficiency of the active Cu component for glycerol hydrogenolysis. Among several supported catalysts, the γ-Al2O3-supported Cu catalysts showed superior performance. The alumina support probably contributes to the glycerol conversion by promoting the formation of reaction intermediates on its acid sites. Compared with a commonly used commercial copper chromite catalyst, Cu/Al2O3 presented much higher activity, selectivity and Cu usage efficiency. The experimental results combined with characterization revealed that unsaturated copper species, such as Cu0 and Cu+, are responsible for the hydrogenolysis of glycerol. It was suggested that the pre-reduction of the Cu species plays a crucial role in the catalytic performance of Cu-based catalysts. Cu on these catalysts is initially in an oxide state. To achieve a decent activity, the catalysts often need to be reduced in situ under a very high H2 pressure47 or to be pre-reduced to generate Cu species that are catalytically active under mild reaction conditions. Zhou et al. eliminated the pre-reduction step while maintaining or further improving the activity and selectivity by adding another metal to prepare M–Cu supported bimetallic catalysts.48 A synergistic effect is observed between Cu and Ag impregnated on γ-Al2O3. The TPR pattern of Cu/Al2O3 displays a peak centered at about 523 K, which corresponds to the reduction of CuO, starting at approximately 473 K and ending at approximately 533 K. The TPR pattern of the CuAg/Al2O3 catalyst contains a broad major peak around 423 K, which can be assigned to the reduction of CuO to metallic copper (Fig. 1). The reduction of the oxide species on this catalyst is completed at approximately 473 K. Therefore, they concluded that a similar reduction occurred under the particular reaction conditions (473 K, 3.6 MPa). The XPS spectrum of as-prepared CuAg/Al2O3, which is calcined at 673 K in air, also shows CuO as the only Cu species. After one reaction cycle at 473 K in the presence of glycerol and hydrogen, the satellite peaks (963 and 943 eV) from the catalyst diminished significantly and the Cu 2p signals were slightly shifted to lower binding energies (952.5 and 932.3 eV), indicating that CuO was mostly reduced (Fig. 2). This confirms that Ag on the CuAg/Al2O3 catalyst lowers the reduction temperature of the copper oxide and makes in situ reduction possible. The H2-TPR analysis shows that Ag oxide on Al2O3 can be completely reduced at a temperature of no more than 393 K. Thus, at 473 K in the presence of hydrogen, silver oxide is easily reduced to metallic Ag, which further facilitates hydrogen spillover and promotes the reduction of Cu oxides in situ. The CuAg/Al2O3 catalyst has high activity and almost 100% selectivity to propanediols under mild reaction conditions and does not need a reduction pretreatment. Compared with Al2O3, the more acidic supports, HY, Hβ and HZSM-5, did not generate catalysts with high activity at the same loading of Cu and Ag. As aforementioned,33–35,40,41,46 it has been observed that irrespective of the method of preparation of the catalysts, when Al2O3 is used as a catalyst support, in most cases glycerol conversion and product selectivity are consistently comparable with other catalysts. Al2O3 not only provides moderate acidic functionality for removal of an –OH group but also has a beneficial effect on the properties of metals supported on it. For example, the reduction of the metals on zeolite supports, such as HY and Hβ, is delayed to a higher temperature when compared with the reduction on Al2O3, which forms a suitable metal-support interaction with the metals.47
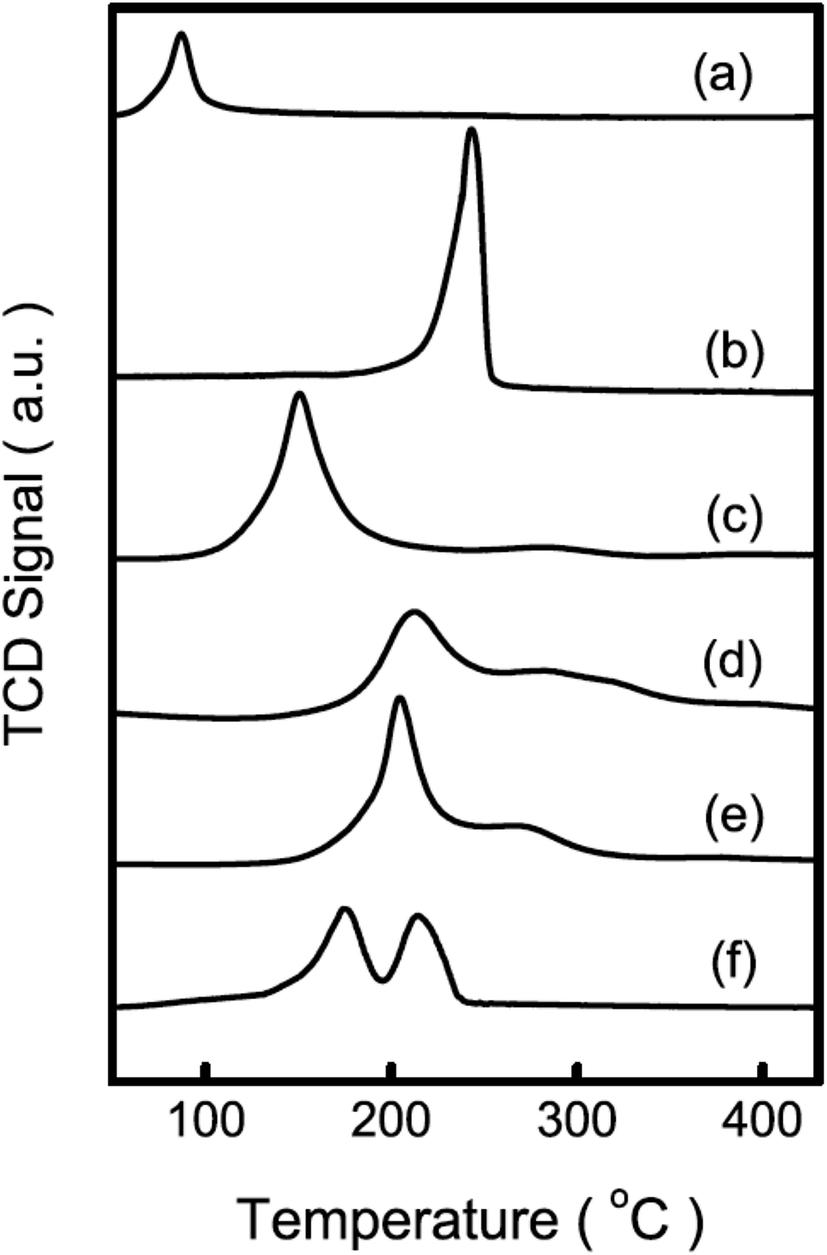 | ||
| Fig. 1 TPR patterns of Cu and other metal impregnated Al2O3 catalysts with various total metal contents (mmol g−1, listed in the first brackets) and molar ratios (listed in the second brackets): (a) Ag/Al2O3 (2.0), (b) Cu/Al2O3 (2.0), (c) CuAg/Al2O3 (2.7) (7:3), (d) CuZn/Al2O3 (2.7) (7:3), (e) CuCr/Al2O3 (2.7) (7:3), and (f) Cu/Al2O3 (1.9) + Ag/Al2O3 (0.8) (Reprinted from ref. 48). | ||
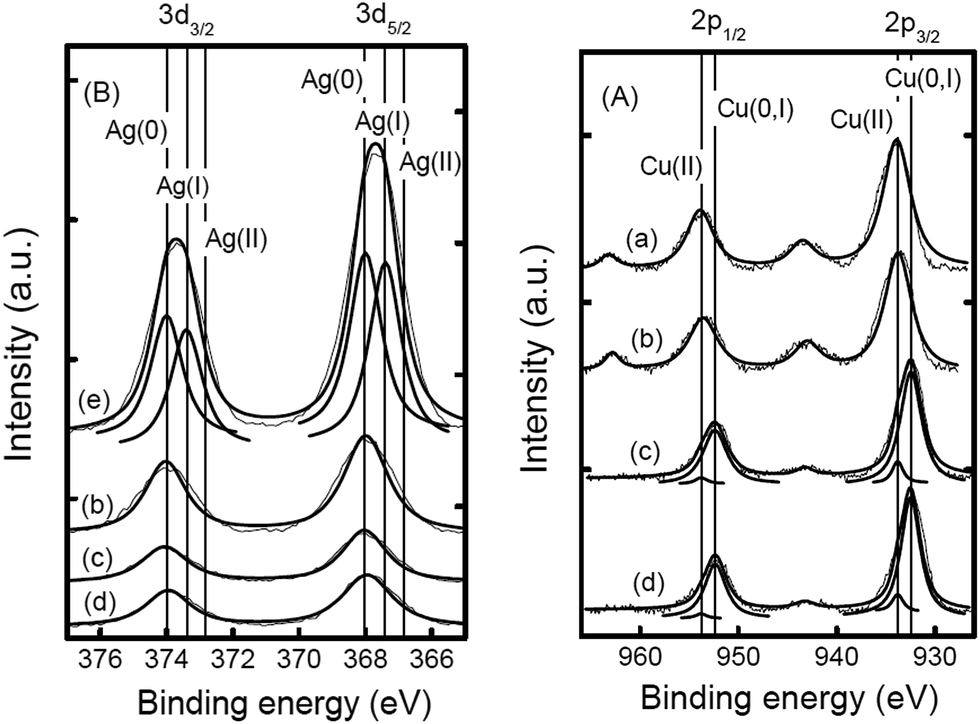 | ||
| Fig. 2 XPS spectra in the binding energy range of (A) Cu 2p and (B) Ag 3d of CuAg/Al2O3 catalysts with various total metal contents (mmol g−1, listed in the first brackets) and molar ratios (listed in the second brackets): (a) Cu/Al2O3 (1.9), (b) as-prepared CuAg/Al2O3 (2.7) (7:3), (c) CuAg/Al2O3 (2.7) (7:3) used at 473 K, (d) CuAg/Al2O3 (2.7) (7:3) reduced in H2 at 473 K, and (e) Ag/Al2O3 (2.0) (Reprinted from ref. 48). | ||
| Catalyst | Preparation method | T (K)/P (MPa,H2)/time (h) | Water (g)/glycerol (g)/catalysta (%) | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|
| 1,2-PD | ||||||
| a With respect to glycerol mass unless particularly stated.b Cu/glycerol (mass ratio).c Evaporation-induced self-assembly. | ||||||
| RANEY® Cu/MgO | Physical mixture + calcination | 453/1.0/6 | 16.86/16.86/2.14 | 48.7 | 83.2 | 49 |
| CuO/CuCr2O4 | Epoxide-assisted sol–gel | 473/4.1/5 | 9.22/13.83/5 | 35 | 90 | 50 |
| CuO/CuCr2O4 | Sol–gel | 483/4.15/10 | 21.66/41.49/5 | 52.0 | 88.0 | 51 |
| CuO/CuCr2O4 | Sol–gel | 403/2.0/4 | 0/18/3.18b | 52.4 | 99.6 | 52 |
| CuO/CuCr2O4 | Facile carbon template | 483/4.15/10 | 46.06/30.71/5 | 51.0 | 97.1 | 53 |
| CuO/CuFe2O4 | Sol–gel | 463/4.1/10 | 9.22/13.83/5 | 47.0 | 92.0 | 54 |
| Cu/boehmite | Aqueous chemical reduction | 473/4.0/6 | 4/16/5 | 77.5 | 92.5 | 55 |
| CuAl2O4 spinel | Sol–gel | 493/5.0/12 | 0/50/2 | 90.0 | 91.0 | 56 |
| CuZnAl2O4 | EISAc | 453/4.0/10 | 4.86/19.43/0.5b | 85.8 | 92.1 | 57 |
CuAl2O4 and Cu–ZnO–ZnAl2O4 catalysts synthesized by sol–gel and evaporation-induced self-assembly (EASI) methods, respectively,56,57 show similarly impressive 1,2-PD yields (>75%), although the structures of these two catalysts are apparently different. For the CuAl2O4 spinel, CuAl2O4 is the only phase when the precursor is calcined at 1073 K. High catalytic activity is attributed to the outstanding reducibility of the Cu species and its strong activity for hydrogen adsorption/desorption according to the analyses of H2-TPR and H2-TPD characteristics. Nevertheless, ZnAl2O4 spinel merely functions as a carrier that improves the chemical and thermal stability of the catalyst, whereas a strong interaction between nano-ZnO and nano-Cu particles enhances the catalyst's performance. The results illustrate that spinel compounds can be effective components of catalysts for glycerol hydrogenolysis. Cu–ZnO–ZnAl2O4 catalysts prepared using a one-pot method via the evaporation-induced self-assembly (EISA) of Pluronic P123 and the corresponding metal precursors exhibited the highest yield (79%) of 1,2-PD with 85.8% glycerol conversion and 92.1% 1,2-PD selectivity at 453 K in 80 wt% glycerol aqueous solution over a 10 h reaction time.57 The high catalytic activity was attributed to the presence of the ZnAl2O4 support, the strong interaction between ZnO and Cu nanoparticles and the small particle size of ZnO and Cu.
Thus, it can be concluded that Cu supported on acidic carriers such as γ-Al2O3 and spinel as well as basic carriers, including MgO and ZnO, are promising catalysts for 1,2-PD production. Highly dispersed Cu0 and the existence of Cu+ are responsible for activating hydrogen and promoting the catalyst stability, whereas activation of glycerol is merely achieved by supports with proper acidity or basicity. Hydrogen spillover, generated by noble metals, promotes the reduction of CuO to Cu and in situ reduction can occur, which results in more Cu species and a higher metallic surface area. Interaction between metal oxides and supports enhances acidity or basicity as well as catalyst stability and thus some multi-metal-oxide involved Cu catalysts are more efficient and attractive. Furthermore, appropriate operation conditions, including reaction temperature, time and pressure as well as catalyst and glycerol concentration, can suppress secondary reactions, leading to an improved 1,2-PD selectivity and better reusability of Cu catalysts.
3.2 Ni and Co-based catalysts
In addition to Cu catalysts, Ni- and Co-containing catalysts as a class of traditional metal catalysts have been applied to glycerol hydrogenolysis and their catalytic results are summarized in Table 4. Similar to Cu catalysts, the Ni and Co catalysts mainly hydrogenate the –OH linked on the terminal carbon of glycerol, thus 1,2-PD is the main propanediol product. RANEY® Ni achieves a 48.5% 1,2-PD yield from pure glycerol under mild reaction conditions of 1 MPa and 463 K.58 However, a very high catalyst loading of 31.25 wt% with respect to glycerol is adopted to obtain a distinct conversion due to the poor dispersion of the Ni component. Dispersion of the Ni component on the supports makes more metal sites available to glycerol molecules; as a result, the catalyst loading is lowered.59–64 Because the addition of Al2O3 can enhance the acid function of the catalyst and promote dehydration of glycerol during hydrogenolysis, Ni/Al2O3–SiO2 catalysts with a high metal dispersion yield a considerable 1,2-PD selectivity of 98% at 2.5 MPa.60 Bimetallic Ni–Cu/Al2O3 catalysts present a better catalytic performance towards 1,2-PD production compared with Ni/Al2O3 and Cu/Al2O3 catalysts due to the synergy of Cu and Ni. Cu promotes the reducibility of NiO and Ni enhances Cu dispersion.63 Moreover, a Ni/Al2O3–CuCr2O4 composite catalyst exhibits a relatively good catalytic performance at low H2 pressure due to the interaction between the metal and support as well as the modest hydrogenolytic activity towards the C–C bonds of Cu species.64 A high conversion (95.1%) and an acceptable 1,2-PD selectivity (85.9%) is achieved on a Ni2P/SiO2 catalyst in a fixed bed reactor. The weak acid sites provided by Ni2P and its electronic and geometrical properties restrict the cleavage of C–C bonds. Moreover, interaction between the P sites in Ni2P and the O atom in glycerol might also favor the activation and cleavage of C–O bonds, resulting in a very high 1,2-PD yield of 81.9% in the hydrogenolysis of glycerol.62| Catalyst | Preparation method | T (K)/P (MPa,H2)/time (h) | Water (g)/glycerol (g)/catalysta (%) | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|
| 1,2-PD | ||||||
| a With respect to glycerol mass unless particularly stated, “-” not mentioned.b 60 wt% glycerol solution, LSHV = 3.0 h−1, molar ratio of hydrogen/glycerol solution = 2:1 with a GHSV of 1060 h−1.c WSHV, 60 wt% glycerol solution, molar ratio of H2/reactant molar = 20.d WSHV, molar ratio of H2/reactant molar = 20. |
||||||
| RANEY® nickel | Alkali-leaching | 463/1.0/20 | 0/8/25 | 63.0 | 77.0 | 58 |
| Ni/AC | Carbothermal-reduction | 473/5.0/24 | 112.5/37.5/1.8 | 63.2 | 77.4 | 59 |
| Ni/SiO2–Al2O3 | Commercial | 473/2.5/8 | -/-5 | 30.0 | 98.0 | 60 |
| Ni/SiO2 | Commercial | 523/6.0/- | -/-/8.5b g | 25.6 | 70.2 | 61 |
| Ni2P/SiO2 | Impregnation | 493/3.0/- | 1.13c h−1 | 95.1 | 85.9 | 62 |
| Ni–Cu/Al2O3 | Sol–gel | 493/4.5/24 | 36.71/1.65/16.6 | 70.5 | 66.9 | 63 |
| Ni/Al2O3–CuCr2O4 | Impregnation | 473/1.5/8 | 0/-/5d | 32.11 | 94.1 | 64 |
| Ni/Al2O3–CuCr2O4 | Impregnation | 473/1.5/20 | 0/-/5d | 46.54 | 95.0 | 64 |
| CoNi | Polyol-synthesis | 493/3.0/7 | 36/4/1.25 | 59.1 | 50.4 | 65 |
| Co–Al alloy | Not mentioned | 473/4.0/15 | 3.06/27.61/32.6 | 100 | 70.1 | 66 |
| CoZnAl2O3-873 K | Co-precipitation | 473/2.0/12 | 36/4/7.5 | 70.6 | 57.8 | 67 |
| CoZnAl2O3-673 K | Co-precipitation | 473/2.0/12 | 36/4/7.5 | 67.7 | 50.5 | 67 |
| Co hexagonal plates | Precipitation | 493/6.0/8 | 76/4/2.5 | 25.8 | 60.02 | 68 |
| Co polyhedrons | Precipitation | 493/6.0/8 | 76/4/2.5 | 33.5 | 47.6 | 68 |
Research related to Co catalysts is less frequently reported in comparison to that on Cu and Ni catalysts. Guo et al.65–68 prepared various Co materials, such as CoNi nanowire, nano-Co plates and Co–Al, and applied them for glycerol hydrogenolysis to prepare 1,2-PD. The anisotropic shape and particle size of CoNi is responsible for the catalytic performance.65 In the case of a CoAl catalyst, the strong synergistic effect between the two metals has a significant influence on the high 1,2-PD yield. The difference in electronegativity between Al and Co generates a highly dispersed Co species and simultaneously enhances the stabilization of species formed.66 Guo et al.67 believed that the pre-reduction temperature had a crucial effect on the oxidation state and dispersion of Co species in Co/ZnO–Al2O3 catalysts. CoO and metallic Co were obtained when the catalysts were reduced at 673 K and 873 K, respectively. However, the Co-673 catalyst performs similar to the Co-873 catalyst and it seems that CoO species are reduced under the reaction conditions since metallic Co is observed on the used sample. A mono-metallic Co catalyst without a carrier also exhibited a certain performance towards 1,2-PD.68 In general, 1,2-PD selectivity on Co-based catalysts is lower than that on Cu- or Ni-based catalysts.
4. Noble-metal catalysts
Noble metals, such as Pt, Ir and Ru, are known to activate hydrogen molecules, therefore monometallic and multimetallic noble-metal species, combined with acidic or basic components, are frequently used in the preparation of metal–acid bifunctional catalysts for the hydrogenolysis of glycerol. Supported Pt on solid acid or base has been applied for propanediol production (Table 5). Checa et al. point out that a Pt–ZnO catalyst exhibited a better catalytic performance than Pt–SnO2, Pt–ZrO2 and Pt–TiO2 due to its stronger metal-support interaction.69 Other metal–ZnO catalysts like Rh–ZnO and Pd–ZnO as well as Au–ZnO were also applied for 1,2-PD production. The conversion decreased in the order Pt > Rh ≫ Pd ≫ Au. The role of the acid and metal sites on product selectivity over Pt/amorphous silico alumina (ASA) catalysts was reported by Gandarias et al.70 Acid sites provided by both ASA and Pt are involved in glycerol dehydration to acetol, whereas subsequent hydrogenation of acetol to 1,2-PD is accomplished by Pt alone (Scheme 3, Route 1). Although Pt promotes hydrogen spillover and restricts the formation of coke, the selectivity to 1,2-PD is as low as 31.9% due to C–C bond cleavage and further hydrogenolysis of 1,2-PD to 1-propanol. Although Pt/solid acid catalysts show poor selectivity, a Pt/solid base catalyst, Pt/hydrotalcite (Mg6Al2(OH)16CO3·4H2O), achieved both high conversion and good selectivity to 1,2-PD following the dehydrogenation–dehydration–hydrogenation route (Scheme 3, Route 2) under mild conditions.71 Similar to Ru/C catalysts, Amberlyst 15 was also used as a co-catalyst to enhance the acidity in a Pt/Nb2O5/Al2O3 catalytic system.72 Pt/Nb2O5/Al2O3 itself has acidity due to the reaction of the Nb precursor with the hydroxyl functionality of the support, but the total acidity is not enough to catalyze the dehydration reaction of glycerol. A physical mixture of a Pt/xNb2O5/Al2O3 catalyst and Amberlyst 15 improved the catalyst performance. A Pt–Sn bimetal catalyst, Pt–Sn/SiO2, also exhibited improved selectivity to 1,2-PD.73 Sn addition enhances the electronic density on the Pt atom and favors polarization of the C–OH bond. On the other hand, ionic Sn species act as ‘‘Lewis acid sites’’ to promote the C–O cleavage to obtain acetol and finally afford 1,2-PD.| Catalyst | Preparation method | T (K)/P (MPa,H2)/time (h) | Water (g)/glycerol (g)/catalysta (%) | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|
| 1,2-PD/1,3-PD | ||||||
| a With respect to glycerol mass unless particularly stated.b 7.7 wt% of Amberlyst 15 was added.c 1,2-PD and 1,3-PD.d 5% total metal loading with Pt/Re molar ratio of 1 (2.6 nm).e WHSV.f Pretreatment by hydrothermal method.g A certain amount of H2SO4 (H+/Re = 1, molar ratio) was added.h 1.5 wt% of HZSM-5 was added (HZSM-5/glycerol).i 6 wt% of HZSM-5 was added (HZSM-5/glycerol).j 1.5 wt% of HZSM-5 and 2.5 wt% of Amberlyst 70 were added.k 0.04 wt% of H2SO4 (H+/Re = 1, molar ratio) was added. | ||||||
| Pt/ZnO | Deposition–precipitation | 453/2.0/12 | 100/6.26/0.48 | 19.0 | 50.0/NM | 69 |
| Pt/ASA | Commercial | 493/4.5/24 | 34.31/8.58/1.94 | 19.0 | 31.9/4.5 | 70 |
| Pt/hydrotalcite | Impregnation | 493/3.0/20 | 20/4/12.5 | 92.1 | 93.0/0 | 71 |
| Pt/Nb2O5/Al2O3 + Amberlyst 15 | Physical mixtures | 413/5.0/10 | 83.69/20.92/3.58b | 78.0 | 95.0/NM | 72 |
| PtSn/SiO2 | Cationic exchange | 498/1.6/2 | 10.4/1.23/40.75 | 16.0 | 84.0c | 73 |
| Pt–Re/Cd | Co-impregnation | 443/4.0/8 | 19.8/0.2/14 μmol Pt | 57.0 | 55.0/15.0 | 74 |
| Pt/commercial WO3 | Impregnation | 453/5.5/12 | 36/4/25 | 4.5 | 14.1/29.9 | 75 |
| Pt/mesoporous WO3 | Impregnation + EISA | 453/5.5/12 | 36/4/25 | 18.0 | 4.1/39.3 | 75 |
| Pt/WO3/TiO2/SiO2 | Co-impregnation | 453/5.5/12 | 36.81/4.09/62.74 | 15.3 | 9.2/50.5 | 76 |
| Pt/W20–Ti80 | Impregnation + EISA | 453/5.0/12 | 36.81/4.09/24.45 | 55.4 | 11.8/26.9 | 77 |
| Pt–HSiW/ZrO2 | Impregnation | 453/5.0/0.09e h−1 | 10 wt%/2 g (cat.) | 24.1 | 16.5/48.1 | 78 |
| Pt/WO3/AlOOH | Successive impregnation | 453/5.0/12 | 3/0.092/109 | 100 | 2/66.0 | 79 |
| Pt/WO3/γ-Al2O3 | Successive impregnation | 453/5.0/12 | 3/0.092/109 | 42.0 | -/19.0 | 79 |
| Pt/WO3/γ-Al2O3f | Successive impregnation | 453/5.0/12 | 3/0.092/109 | 89.0 | 20.0/35.0 | 79 |
| Ir–ReO/SiO2 | Impregnation | 393/8.0/36 | 1/4/3.75 | 81.0 | 5.0/47.0 | 24 |
| Ir–ReOx/SiO2 | Impregnation | 393/8.0/4 | 1.98/4.02/3.73g | 21.7 | 6.7/58.9 | 25 |
| Ir–ReOx/SiO2 + HZSM-5 | Physical mixtures | 393/8.0/24 | 16/4/3.75h | 58.8 | 44.7/9.3 | 80 |
| Ir–ReOx/SiO2 + HZSM-5 | Physical mixtures | 393/8.0/24 | 4/1/15i | 51.6 | 6.3/53.3 | 80 |
| Ir–ReOx/SiO2 + HZSM-5 + Amberlyst 70 | Physical mixtures | 393/8.0/24 | 16/4/3.75j | 69.7 | 44.4/8.0 | 80 |
| Ir–ReOx/SiO2 + H2SO4 | Physical mixtures | 393/8.0/24 | 16/4/3.75k | 61.1 | 43.2/9.2 | 80 |
A few Pt catalysts containing Re74 or WOx species75–79 have the potential to produce 1,3-PD. The selectivities of 1,2-PD and 1,3-PD on Pt–Re/CNTs (carbon nano tubes) catalysts are closely related to the Pt/Re ratio.74 The metal particle size decreased continuously with an increase in surface Re. The Pt–Re/CNTs catalyst with a smaller particle size favored the scission of the primary C–O bond, while the one with a larger particle size is active in the scission of the secondary C–O bond of glycerol. More 1,3-PD is produced over the catalyst with a larger particle size of around 4.9 nm, although the 1,3-PD selectivity is still less than 20%. A higher 1,3-PD selectivity (39.3%) is obtained on a supported Pt/mesoporous tungsten trioxide (m-WO3) catalyst.75 The m-WO3 reduced Pt components more easily compared with the commercial WO3 support due to the presence of a large number of oxygen vacancies. It is believed that hydride (H−) attacks the 2-position of alkoxide to break the C–O bond and to form the 3-hydroxypropoxide carbanion, which is subsequently attacked by H+ to form 1,3-PD. The higher dispersion of Pt and easier reducibility of m-WO3 promote the formation of protons as well as hydride and thus the molar ratio of 1,3-PD to 1,2-PD was more than 4 times that produced over the Pt/commercial WO3. Another WO3-containing catalyst, Pt/WO3/TiO2/SiO2, showed a better 1,3-PD selectivity of 50.5% under similar reaction conditions to the Pt/m-WO3 catalyzed reaction. The authors attributed this to the synergistic functions provided by TiO2, SiO2 and WO3.76 TiO2 improves the dispersion of platinum due to the strong interaction between Pt and TiO2, whereas WO3 plays a key role in generating Brønsted acid sites, both enhancing the total acid amount and acid strength. SiO2 contributes to a high surface area and the stronger interaction between silica and glycerol, due to the hydrophilic nature of silica, which results in a higher glycerol absorption and rapid desorption of the product from the catalyst surface. A Pt/Wn–Ti100−n catalyst prepared by the modified evaporation-induced self-assembly (EISA) method achieved 14.9% yield of 1,3-PD.77 It was suggested that dispersion of tungsten oxide in the W–Ti composite oxide and redox of W6+/W5+ were responsible for the improvement in 1,3-PD production. The introduction of tungstic heteropoly acids (HPAs) in Pt/ZrO2 also improved the 1,3-PD yield due to the enhanced acidity, especially the Brønsted acid strength.78 The 1,3-PD selectivity on Pt/HPAs/ZrO2 catalysts was arranged in the order Pt/H4SiW12O40 (HSiW)/ZrO2 > Pt/H3PW12O40 (HPW)/ZrO2 > Pt/H3PMo12O40 (HMoW) at 453 K, which is similar to the sequence of the amount of Brønsted acid. A high 1,3-PD yield of 66% at 100% glycerol conversion in an aqueous solution was achieved on a Pt/WOx/AlOOH (boehmite) catalyst after the hydrothermal treatment.79 The Al–OH component on the boehmite surface, in combination with the WOx species, was responsible for the excellent catalytic performance towards 1,3-PD. Initially, glycerol adsorbs onto the boehmite surface and one of its primary OH groups reacts with an AlOx group to form the Al-alkoxide species (HOCH2CH(OH)CH2OAlOx). Then, the WOx species promotes the dehydration of the secondary OH group in glycerol alkoxide to generate a secondary carbocation intermediate. Finally, the hydride species on WOx from spillover attacks the carbocation intermediate, followed by the hydrolysis of glycerol alkoxide to 1,3-PD. The good dispersion of Pt and WOx on the boehmite surface and the strong interactions between the support and Pt as well as WOx resulted in excellent reusability of Pt/WOx/AlOOH, as demonstrated by 10-times cycles.
Hydrogenolysis of glycerol to 1,3-PD is more challenging than to 1,2-PD since most catalysts are active in dissociating the primary hydroxyl of glycerol. In addition to the aforementioned Pt catalysts, rhenium-oxide-modified supported iridium nanoparticles on silica catalysts, Ir–ReO/SiO2, provided new possibilities to obtain higher selectivity to 1,3-PD.24,25,80 A high selectivity of 47% to 1,3-PD at 81% conversion of glycerol was observed over Ir–ReOx/SiO2 (Re/Ir = 1), while both Ir/SiO2 and ReOx/SiO2 catalysts showed very low catalytic activity and selectivity to 1,3-PD.24 The synergy between Ir and Re enabled the hydrogenolysis of glycerol to 1,3-PD. Based on the combined characterization results and related experiments, it was suggested that 1,3-PD is produced by the attack of active hydrogen species on iridium metal on 1-glyceride species formed on the oxidized rhenium cluster. ICP analysis of the reaction solution after filtration of the catalyst showed no appreciable leaching of Ir or Re. The Ir–ReOx/SiO2 catalyst could be reused at least three times without a change in the reaction rate or the selectivity. The mechanism of the hydrogenolysis of glycerol to 1,3-PD over the Ir–ReOx/SiO2 catalyst was discussed.25 The conversion in the glycerol hydrogenolysis increased with an increasing amount of Re up to Re/Ir = 2, and the high selectivity to 1,3-PD (ca. 60%) was almost independent of the Re amount. The average size of an Ir metal particle gradually decreased as the amount of Re increased. The Ir metal surface was partially covered with ReOx clusters, regardless of the Re amount. The reaction order on H2 pressure over Ir–ReOx/SiO2 (Re/Ir = 1) was one, suggesting that one active hydrogen species was produced from one hydrogen molecule. A low glycerol reaction order indicated a strong interaction between glycerol and the catalyst surface. These reaction trends and characterization results supported the direct reaction mechanism for the formation of 1,3-PD from glycerol via 2,3-dihydroxypropoxide, as discussed in the section on reaction mechanisms.
Hydrogenolysis of glycerol over Ir–ReOx/SiO2 was promoted by the addition of various types of acids such as zeolites, silica–alumina, ion-exchange resin and sulfuric acid. Among all the solid acids tested, Amberlyst 70 and H-ZSM-5 were selected as promising additives since their reaction systems exhibited enhanced glycerol conversion and 1.3-PD yield. Ir–ReOx/SiO2 + Amberlyst 70 is very effective for obtaining high activity. The promoting effect of Amberlyst 70 is even larger than that of H2SO4 under the same conditions. However, the Ir–ReOx/SiO2 + Amberlyst 70 system had poor reusability since the ion-exchange resin was burned during the recycling process. Considering the reusability, H-ZSM-5 is a more suitable co-catalyst due to the stabilization of H-ZSM-5 at high temperatures and the good promoting effect. The Ir–ReOx/SiO2 + H-ZSM-5 system showed kinetic and selectivity trends under various reaction conditions similar to those of Ir–ReOx/SiO2 + H2SO4. The hydroxorhenium site (Re-OH) in the ReOx cluster was the adsorption site of glycerol and the addition of acids benefited the stabilization of the structure, which resulted in a better activity than in the neutral system.80
4.1 Ru-based catalysts
Ruthenium (Ru) is a well-known active component for activating hydrogen molecules, and Ru-based catalysts are widely used in the hydrogenolysis of glycerol. The reactions and processes for glycerol hydrogenolysis on Ru-based catalysts have been well covered in the review by Tomishige et al.13 In this study, several typical and recent investigations about Ru-based catalysts are introduced (Table 6).| Catalyst | Preparation method | T (K)/P (MPa,H2)/time (h) | Water (g)/glycerol (g)/catalysta (%) | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|
| 1,2-PD | ||||||
| a With respect to glycerol mass unless particularly stated.b 7.17 wt% of Amberlyst 15 was added (Amberlyst 15/glycerol).c 140 μmol H+ was added.d 0.4 mmol H+ was added. | ||||||
| Ru/AC-Ox | Impregnation | 453/8.0/24 | 92.03/10.22/4.4 | 42.0 | 8.0 | 81 |
| Ru/C + Amberlyst 15 | Physical mixtures | 393/8.0/10 | 16.74/4.18/3.58b | 12.9 | 55.4 | 18 |
| Ru/C + Amberlyst 70 | Physical mixtures | 453/8.0/10 | 16.74/4.18/3.58c | 48.8 | 70.2 | 82 |
| Ru/C + DAE26-4 | Physical mixtures | 473/8.0/4 | 8/1.92/0.02 mmol Rud | 23.0 | 33.0 | 83 |
| Ru/C + Amberlyst 70 | Physical mixtures | 453/8.0/4 | 5/1.2/0.02 mmol Rud | 54.0 | 2.0 | 83 |
| Ru/SiO2 | Impregnation | 513/8.0/5 | 0/153.96/0.58 | 21.7 | 60.5 | 84 |
| Ru/SiO2 | Impregnation | 513/8.0/5 | 29.14/116.57/0.6 | 18.0 | 90.2 | 85 |
| Ru/CeO2 | Impregnation | 453/5.0/10 | 2.52/0.63/7.97 | 85.2 | 62.7 | 86 |
| RuCaZn/hydrotalcite | Impregnation | 453/2.5/18 | 41.85/10.46/2.87 | 58.5 | 85.5 | 87 |
| RuCu/TMG-BEN | Impregnation + ion-exchange | 503/8.0/18 | 1/0.46/0.15 mmol Ru | 100 | 85.4 | 88 |
| Ru–Cu/ZrO2 | Co-impregnation | 453/8.0/24 | 0.23/0.35/14.46 | 100 | 83.5 | 89 |
| Ru–Re/ZrO2 | Impregnation + precipitation | 433/8.0/8 | 6.58/4.39/3.42 | 56.9 | 47.2 | 90 |
The catalytic conversion of glycerol to 1,2-PD over Ru-supported active carbon (AC or C) has been investigated.81 Ru/AC catalysts present catalytic activity towards the hydrogenolysis of glycerol; however, the selectivity toward 1,2-PD is very low due to a low density of acid groups. Activated carbon treated by nitric acid showed enhanced acidity due to the generation of oxygenated groups (–COOH groups). Thereby, Ru(Cl)/AC-Ox prepared from oxidized carbon (AC-Ox) was more active than Ru(Cl)/AC due to the higher acidity (–COOH groups), but it still exhibited a low 1,2-PD selectivity of only 8%. Moreover, some of the acidic groups, such as carboxylic, lactonic and anhydride groups, decompose during the reduction of the catalysts at 523 K, indicating the poor stability of the acid sites. The combination of Ru/AC with acid catalysts is effective for simultaneous dehydration and hydrogenation.18,82,83 H2SO4 and HCl, as liquid acid catalysts, in combination with Ru/AC are not good choices due to low dehydration activity and poor 1,2-PD selectivity.18 Solid acid resins are much preferred when combined with Ru/AC in glycerol hydrogenolysis. Amberlyst 15 has poor thermal stability and is prone to decompose when the catalyst is used at temperatures higher than 393 K. Sulfur compounds, including SO2 and H2S, were generated by thermal decomposition of the resin and have a poisoning effect on Ru/C, which resulted in a decline in activity. Another type of polystyrene-divinylbenzene resin called Amberlyst 70 showed a higher thermal resistance and the Ru/C + Amberlyst 70 system exhibited a better performance than Amberlyst 15 + Ru/C at a higher reaction temperature.82 However, Amberlyst 70 also generated SO2 and H2S when the operation temperature exceeded 463 K. Compared with Amberlyst 70, an alkysulfonic resin named DAE26-4, which was obtained by the co-polymerization of 2-acrylamido-2-methyl-propansulfonic acid with N,N-dimethylacrylamide and 4% (mol%) ethylene dimethacrylate (the cross-linker), was more thermal resistant and thus could be used at 473 K. The Ru/AC + DAE26-4 systems, however, exhibited relatively low conversion.83
Ru supported that solid acids, such as γ-Al2O3, SiO2 and ZrO2, exhibit higher thermal resistance than observed with external acids as co-catalysts for Ru/AC.84 Both the Ru precursors (such as RuNO(NO3)3 and Ru/Si(NO3)) and the acid support had great influence on the Ru species' dispersion, particle size, reduction temperature and acidity (acid strength and acid density). Catalysts that are too acidic are prone to generate large amounts of propanols produced from over hydrogenolysis of 1,2-PD. RuNO(NO3)3-prepared Ru/SiO2 promotes the hydrogenation of acetol and prevents the excessive hydrogenolysis of 1,2-PD due to the catalyst's lower acidity, indicating that moderate acid sites are desirable to promote glycerol dehydration.84,85
In addition, Ru-supported basic oxides, such as Ru/CeO2, Ru/La2O3 and Ru/MgO, were also applied for the hydrogenolysis of glycerol.86 Compared with MgO or La2O3, Ru particles are well-dispersed on the surface of CeO2. CeO2 is also stable under hydrothermal conditions and the basicity of CeO2 is moderate. As a result, the Ru/CeO2 catalyst exhibited both high activity and high selectivity to 1,2-PD.
Hydrotalcite with a layered double hydroxide structure is known as a weak solid base. Ru/hydrotalcite modified by Ca and Zn exhibited a high 1,2-PD selectivity of 85.5% at a low temperature and hydrogen pressure.87 Addition of Ca decreased the Ru particle size from 20–50 nm to 5–10 nm and thus the smaller particle size of Ru was related to the increase in glycerol conversion during hydrogenolysis.
Ru species exhibit high catalytic activity in the conversion of glycerol but are also active in the cleavage of the C–C bond. Ru–M bimetallic catalysts exhibit synergistic effects between Ru and the second metal in catalyzing the reaction and thus give a better catalytic performance than the corresponding monometallic catalysts.88–90 Ru–Cu/TMGL prepared by supporting Ru–Cu on 1,1,3,3-tetramethylguanidinium lactate ionic liquid (TMGL) exhibited a considerable 1,2-PD yield of 85%.88 Similarly, addition of Cu to Ru/TMG-BEN restrained the cleavage of the C–C bond and the selectivity to 1,2-PD increased to 85.4% when the molar ratio of Ru/Cu was 3/1. Ru–Cu/ZrO2 showed the highest glycerol conversion and 1,2-PD selectivity of 100% and 84.0%, respectively, exhibiting higher activity than Ru–Cu/SiO2, Ru–Cu/TiO2, Ru–Cu/Al2O3, Ru–Cu/NaY and Ru–Cu/HY.89 Re also serves as a second metal with Ru. Re species in Ru–Re/ZrO2 promote the dispersion of Ru and prevent the aggregation of Ru metal particles during the reaction; moreover, rhenium oxide species also serve as solid acids that promote the glycerol dehydration step.90 Therefore, the synergistic effect between Ru and Re promotes glycerol conversion and 1,2-PD selectivity compared with Ru monometallic catalysts.
4.2 Ag-based catalyst
Ag is a relatively cheap noble metal component. Ag-based catalysts have been synthesized and used in propylene epoxidation.91–95 Recent research illustrates that Ag-based catalysts can also be applied in glycerol hydrogenolysis.96,97 Unlike other noble metal catalysts, Ag-based catalysts are prone to achieving a high 1,2-PD selectivity of 90% or more. Silver incorporated octahedral molecular sieve (OMS-2) catalysts for the hydrogenolysis of glycerol have been studied in both batch and continuous modes of operation.96 A high conversion of glycerol (about 65%–70%) with about 90% selectivity toward 1,2-PD was obtained within 8 h under hydrogen pressure in a batch reactor. The glycerol conversion was examined in a fixed bed reactor for up to 150 h, which demonstrated that the catalyst was very stable. A series of γ-Al2O3-supported silver catalysts (Ag/Al2O3), prepared with various Ag loadings and calcination temperatures, were also used to convert glycerol to 1,2-PD.97 A catalyst with 2 mmol Ag per gram Al2O3 calcined at 673–773 K presented the highest activity (46%) and selectivity (96%) at 493 K, where glycerol/Ag (molar ratio) = 100/2, initial H2 pressure = 1.5 MPa and reaction time = 10 h. However, catalyst deactivation was observed. Sintering due to the operation conditions was responsible for the activity loss. Deactivation resulting from coking was minimal under the reaction conditions. The deactivated catalyst could be fully regenerated with calcination in air, which re-dispersed the sintered Ag particles and also burned off the coke formed.5. Some special process features
In addition to the significant impact of catalysts on the reaction performance, other technological aspects, such as hydrogen sources, reaction solvents, reactor types, feeding processes and reaction temperature, time and pressure, also play important roles in dictating the catalytic activity and selectivity.In the aforementioned glycerol hydrogenolysis reactions, molecular hydrogen is consumed as a feedstock. Due to the low solubility of hydrogen in glycerol/water solutions, H2 availability to active sites is a dominant factor in the molecular hydrogenation process.70,98 In order to reach acceptable conversions, high hydrogen pressures are required in liquid-phase reactions.70,98–100 At present, most of the available hydrogen gas is produced from fossil fuels by energy intensive processes.99 This presents considerable hazards and high costs associated with the purchase, transport, storage and usage of the high pressure hydrogen. Therefore, developing a process of in situ generation of hydrogen has been considered as a promising solution to avoid the inherent drawbacks of the molecular-hydrogen process.
Hydrogen production from glycerol reforming has been reported.101–103 Part of glycerol initially combines with water to generate hydrogen and CO2 in the reforming step: C3H5(OH)3 + 3H2O → 3CO2 + 7H2. Hydrogen is then consumed in the hydrogenolysis of glycerol, leading to an overall conversion of glycerol to 1,2-PD, CO2 and water.70 Nevertheless, the results published to date on the combination of liquid-phase glycerol hydrogenolysis and glycerol reforming are still inferior to the best results reported under H2 pressure. Hydrogen production from several monohydric alcohols, such as 2-propanol,104–106 methanol104 and ethanol,106 has been reported to occur through catalytic transfer hydrogenation (CTH) processes, in which hydrogen is transferred from a hydrogen donor molecule to an acceptor. 2-Propanol proved to be more effective as a hydrogen source for the glycerol hydrogenolysis process under N2 pressure to yield 1,2-PD compared to glycerol aqueous-phase reforming.105 In the reaction containing 2-propanol as the hydrogen production source, PdO in the calcined catalyst was first reduced by 2-propanol to afford metallic Pd at 453 K. Then, the Pd0 sites catalyzed the dehydrogenation of 2-propanol to generate H2.106 A drawback of the 2-propanol process is that glycerol hydrogenolysis with 2-propanol is more sensitive to active site deactivation by coke deposition than glycerol hydrogenolysis with molecular hydrogen.105 Besides alcohols, formic acid has also served as a hydrogen donor.98,104,107,108 Gandarias et al.104 compared formic acid with 2-propanol and methanol as hydrogen donor molecules in the glycerol hydrogenolysis process. They found that the best results, with respect to glycerol conversion and 1,2-PD selectivity, are achieved when formic acid serves as a hydrogen donor; the next best results are achieved using 2-propanol. Every molecule of 2-propanol or formic acid generates two hydrogen atoms, but formic acid gives a higher hydrogen efficiency (with around 72% of the hydrogen atoms formed converting to 1,2-PD) than 2-propanol (30%) because part of the propene coming from acetone dehydration is hydrogenated to propane, which consumes hydrogen atoms to some extent. Hydrides are formed from formic acid decomposition and a possible hydrogenolysis process was proposed.107 Initially, formic acid adsorbs dissociatively to afford the formate species and an adsorbed hydrogen atom and then those formate species dissociate to afford gaseous CO2 and another adsorbed hydrogen atom. Since 1,3-dihydroxyisopropoxide adsorbed on an acid site near a metal site has an adsorbed hydrogen atom, the hydride attacks the C–O bond of the alkoxide and finally, hydrolysis of the reduced alkoxide gives 1,2-PD (Scheme 6). Besides dehydrogenation (HCOOH → H2 + CO2), dehydration (HCOOH → H2O + CO) is another pathway with formic acid (FA) as a hydrogen donor, in which the selectivities for the two possible pathways can be controlled by the choice of catalyst.108 When the catalysis involves a low molar ratio of FA to glycerol (1/2), both glycerol conversion and 1,2-PD selectivity are low due to the lack of H2, whereas an excessively high ratio (3/1) results in the conversion of 1,2-PD to n-propanol. Thus, the highest selectivity (95%) for 1,2-PD is obtained with 1 molar ratio of FA to glycerol.
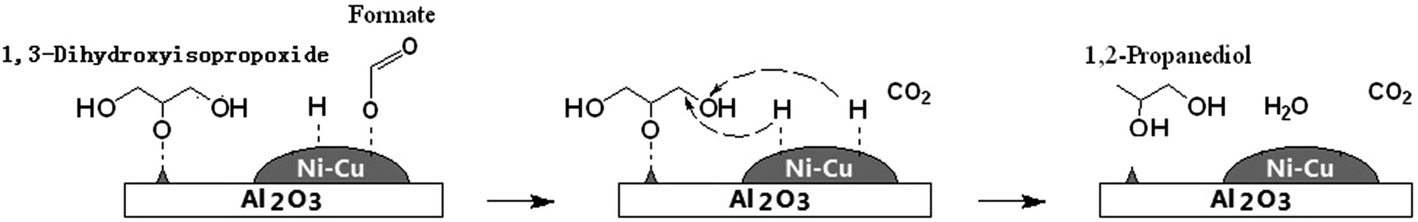 | ||
| Scheme 6 Model structure of the transition states of the hydride attack on the adsorbed 1,3-dihydroxyisopropoxide, under an inert atmosphere, with formic acid as the hydrogen donor molecule over a Ni–Cu/Al2O3 catalyst.107 | ||
Gandarias et al. observed that the hydrogen donor and glycerol compete for the same active sites.105 The number of hydrogen donor molecules should be optimized to balance the positive effect of the higher supply of active hydrogen and the negative effect of the higher competition for active sites.104 In addition, the reaction mechanism is different, depending on whether the origin of the active hydrogen is a molecular hydrogen or a hydrogen donor molecule.105 Acetol, which can be generated by glycerol dehydration on a Cu–Ni/Al2O3 catalyst under molecular H2, cannot be formed using the hydrogen donor,104 indicating the difference in nature between the two mechanisms with different hydrogen sources. Under H2 pressure, glycerol is first dehydrated to acetol, which subsequently undergoes a hydrogenation process to give 1,2-PD109 (Scheme 3, Route 1). When hydrogen atoms are produced from 2-propanol dehydrogenation, glycerol is directly converted to 1,2-PD through intermediate alkoxide formation (Scheme 7).
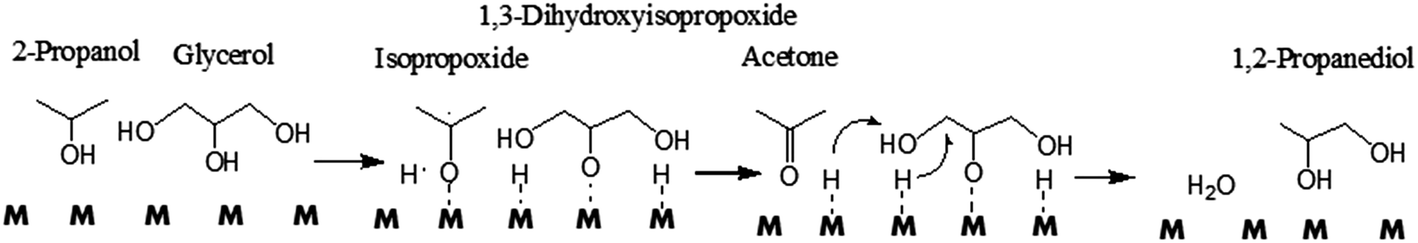 | ||
| Scheme 7 Direct glycerol hydrogenolysis to 1,2-PD using 2-propanol as a hydrogen donor molecule.104 | ||
The solvent is another key factor, impacting catalytic performance. Production of 1,2-PD from glycerol hydrogenolysis has been carried out in a bio-ethanol solvent over a small amount of Rh-promoted Cu/solid-base catalyst.110 The conversion of glycerol increased from 69.3% to 77.8%, 81.8% and 84.0% when the amount of bio-ethanol in the solvent increased from 60% to 70%, 80% and 90%, respectively, under moderate reaction conditions of 2.0 MPa H2 and 353 K. The conversion of glycerol and the selectivity to 1,2-PD in a pure ethanol solvent over the optimized RhCu/MgAlO reached 91.0% and 98.7%, respectively. These results showed that ethanol was a good solvent for the hydrogenolysis of glycerol for higher selectivity and higher conversion of glycerol. The stronger adsorption of water on the surface of the catalyst hindered the accessibility of glycerol to the catalyst surface.111 These excellent results could also be attributed to the higher concentration of hydrogen in the ethanol-containing solvents, since the solubility of hydrogen in ethanol is higher than that in water.
Some efforts have been dedicated towards the modeling and optimization of technological processes for glycerol hydrogenolysis.11,16,87,112–115 A two-step reaction process has been developed using a copper chromite catalyst. The first step was to produce acetol through glycerol reactive distillation at 473 K and 0.65 bar, whereas the second step was to hydrogenate acetol to form 1,2 PD at 473 K and 13.8 bar hydrogen pressure.27 A vapor-phase hydrogenolysis of glycerol has been reported using Cu/ZnO/Al2O3 and Cu/ZnO/ZrO2 catalysts at reaction temperatures of 513–573 K and near-ambient hydrogen pressure.116 Liquid-phase glycerol hydrogenolysis has also been studied in special reactors, and the impacts of various process parameters on conversion and selectivity have been compared for batch and continuous operations.113 All these results were useful for predicting the laboratory reactor performance and for the design of commercial-scale trickle-bed systems (Table 7).
| Catalyst | Atmosphere/solvent/hydrogen donor | T (K)/P (MPa,H2)/time (h) | Total vol. (mL)/Gly. concentration (wt%)/Cat. (g) | Conv. (%) | Select. (%) | Ref. |
|---|---|---|---|---|---|---|
| 1,2-PD | ||||||
| a Catalyst/g of glycerol.b Ex situ reduced at 923 K and then in situ reduced at 723 K in a H2–N2 atmosphere.c Glycerol/2-propanol = 1/1.5 (molar ratio).d Glycerol/Cu (molar ratio).e Catalyst/mL of glycerol.f Evaluated on fixed-bed reactor with liquid flow rate of 30 mL h−1 for 800 h. | ||||||
| Pt/ASA | N2/water/glycerol | 493/4.5/24 | 41/20/0.166a | 26.6 | 35.3 | 70 |
| Cu–NiAl2O3b | N2/water/formic acid | 493/4.5/16 | 25/20/0.87 | 62.0 | 85.0 | 98 |
| Cu–Ni/Al2O3 | N2/water/formic acid | 493/4.5/10 | 20/20/0.5 | 33.5 | 89.5 | 104 |
| Cu–Ni/Al2O3 | N2/water/2-propanol | 493/4.5/24 | 41/4/0.166a | 60.4c | 64.6 | 105 |
| Pd/Fe2O3 | Inert/2-propanol/2-propanol | 453/0.5/24 | 25/12/0.6 | 100 | 94.0 | 106 |
| Cu–Ni/Al2O3 | N2/water/formic acid | 493/4.5/16 | 135/4/0.9 | 47.5 | 78.1 | 107 |
| Cu/ZrO2 | N2/water/formic acid | 473/0.5/18 | 10.36/4.4/10d | 97.0 | 95.0 | 108 |
| Cu–Rh/Mg–Al | H2/ethanol/- | 453/2.0/10 | 7.21/75/1 | 91.0 | 98.7 | 110 |
| Cu/Al2O3 | H2/water/- | 478/2.0/23 | 20/75/0.99 | 88.7 | 94.3 | 112 |
| Cu–Cr–Ba | H2/2-propanol/- | 493/5.2/5 | -/20/0.01e | 34.0 | 84.0 | 113 |
| Cu–Cr–Ba | H2/2-propanol/- | 493/4.0/10f NL h−1 | -/20/23 | 65.0 | >90.0 | 113 |
In addition, the process parameters (e.g. reaction temperature, time and pressure) also play important roles in dictating catalytic activity and selectivity. The reaction temperature exerts a significant impact on the reaction performance. In most cases, increasing the reaction temperature accelerates the conversion of glycerol, whereas the selectivities to propanediols exhibit a volcano-like trend with a maximal value achieved at a moderate temperature. For instance, in the reactions that are catalyzed by Cu/SiO2,30 Cu–Cr,33 Cu/Al2O3,35 Cu/ZnO,38 RANEY®-Cu/MgO49 and Ni/SiO2–Al2O3,60 glycerol conversion increases continuously from 393 K to 513 K and even approaches 100% at a higher temperature, whereas the 1,2-PD selectivity increases initially and then decreases with an increase in the reaction temperature. A dramatic decrease in the 1,2-PD selectivity from 80%–90% to 10%–30% is observed from 473 K to 523 K when the reaction is carried out on Cu/Al2O3 and Ni/SiO2–Al2O3. Similar to Cu- and Ni-based catalysts, noble metal catalysts, such as Ru–Cu/bentonite,88 Ru–Re90 and Ag/Al2O3,91 are sensitive to temperature during the catalytic hydrogenolysis of glycerol. Since 1,3-PD also converts into lower alcohols under harsh conditions, excessively high reaction temperatures result in poor 1,3-PD selectivity.78 Because the catalytic hydrogenolysis of glycerol is an exothermic reaction, a relatively higher reaction temperature is necessary to provide adequate activation energy. However, too much heat favours excessive breakage of the C–O and C–C bonds, which leads to an increase in the lower alcohols. Moreover, the increase in reaction temperature will accelerate the coking rate of the catalysts. Reaction time and hydrogen pressure also have a significant influence on the hydrogenolysis process. A longer reaction time tends to afford a greater 1,2-PD yield, since the process from glycerol to 1,2-PD is a multi-step cascade reaction and needs some time to complete the conversion.36,57 Similarly, glycerol conversion increases with increasing hydrogen pressure. This is because the concentration of hydrogen in the liquid phase increases with pressure, which promotes H2 adsorption on the catalyst surface and facilitates the dissolution of hydrogen in the solution. Moreover, hydrogen is important in maintaining catalyst activation, probably by reducing the oxidized metal in the uncompleted catalysis cycle or by diminishing the adsorbed species on the catalyst surface by hydrogenolysis.49,85,89,99
6. Conclusions and outlook
Catalytic hydrogenolysis of glycerol to propanediol follows different reaction routes, depending on the fundamental properties of the catalytic reaction systems. Among the three typical reaction routes, the dehydration–hydrogenation route and the dehydrogenation–dehydration–hydrogenation route mainly work towards 1,2-PD production, whereas the recently proposed direct-hydrogenolysis route leads to 1,3-PD production. The metal and acid/base properties of the catalysts are the key factors that determine the reaction routes and the product distributions. To obtain high propanediol yields, catalytic hydrogenolysis of glycerol should be controlled to dissociate only one hydroxyl and to avoid cleavage of C–C bonds and over-cleavage of the C–O bond. Therefore, precise control of the bifunctional catalyst properties, including the hydrogenation/dehydrogenation function of the metal species and the hydrolysis function of the acid/base species, is of critical importance. In addition to the selection of metals and acidic/basic solid oxides as the key components to construct the catalysts, efficient preparation methods and precise modulation of the catalyst using modifiers or additives are important. According to the type of metal components, the catalysts used for this reaction are typically classified into two categories: transition metal catalysts and noble metal catalysts. Transition metal catalysts have been intensively investigated with respect to the hydrogenolysis of glycerol, and Cu, Ni, and Co are the most frequently adopted metal components. These catalysts, especially Cu-based catalysts, generally exhibit good 1,2-PD selectivity. Recent efforts using Cu-based catalysts in the hydrogenolysis of glycerol have made progress in improving the efficiency of the active metal components, the catalytic activity and the catalyst stability through adoption of efficient preparation methods and precise modulation techniques. Noble metal catalysts prepared with Ru, Pt, Ir and Ag species in combination with acidic or basic components are widely used in the hydrogenolysis of glycerol. These catalysts are usually more active than transition metal catalysts, but only a few Pt-based catalysts, Ru–Cu catalysts and Ag-based catalysts achieve high selectivity to 1,2-PD. However, several noble-metal catalysts show a promising potential in synthesizing the more valuable 1,3-PD. Several types of Pt catalysts containing Re or WOx species have the potential to produce 1,3-PD. In addition, rhenium-oxide-modified supported iridium nanoparticles on silica catalysts, Ir–ReO/SiO2, provide new possibilities for obtaining higher selectivity to 1,3-PD. In general, for 1,2-PD production, Cu-based catalysts should be specified as the most promising hydrogenolysis catalysts due to their excellent 1,2-PD selectivity, inexpensive components and good activity under mild reaction conditions. Pt and Ir catalysts are promising with respect to catalyzing the more challenging hydrogenolysis of glycerol to 1,3-PD. In addition to the metal species, acid or base supports or co-catalysts also play crucial roles in determining catalyst activity. Except for a few reactions that contain physically blended metal and acid catalysts, most of the catalytic systems adopt metal–metal oxide compounds as the catalysts. Metal oxides, such as Al2O3, SiO2, ZrO2, ZnO and MgO, not only provide acidic or basic functionality for the removal of an –OH group but also act as supports to disperse the metal species. In addition to selecting suitable metal oxides to complex with metals, precise preparation and modulation of the catalysts are of great importance to ensure bifunctionality, i.e., acidic or basic functionality for the removal of an –OH group and oxidation–reduction functionality for addition of hydrogen. The achievements summarized herein serve as the basis for the development of an economically viable commercial production of propanediols from glycerol, although further developments with respect to both catalyst preparation techniques and reaction processes are needed.Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (Grant 21173027, Grant 21203015) and the State Key Laboratory of Fine Chemicals at Dalian University of Technology for supporting the catalyst characterization in this study under Grant KF1109.References
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484 CrossRef CAS PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed.
- H. J. Liu, D. H. Liu and J. J. Zhong, Biotechnol. Prog., 2003, 19, 1615 CrossRef CAS PubMed.
- H. R. Jin, H. Y. Fang and J. Zhug, Biotechnol. Lett., 2003, 25, 311 CrossRef CAS.
- H. Djelal, A. Amrane, F. Lahrer and G. Martin, Appl. Microbiol. Biotechnol., 2005, 69, 341 CrossRef CAS PubMed.
- G. Chen and S. Yao, BioMed Res. Int., 2013, 2013, 467263 Search PubMed.
- C.-H. Zhou, J. N. Beltramini, Y.-X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527 RSC.
- D. T. Johnson and K. A. Taconi, Environ. Prog., 2007, 26, 338 CrossRef CAS PubMed.
- A. Martin, U. Armbruster, I. Gandarias and P. L. Arias, Eur. J. Lipid Sci. Technol., 2013, 115, 9 CrossRef CAS PubMed.
- M. Schlaf, Dalton Trans., 2006, 4645 RSC.
- Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2011, 1, 179 CAS.
- Y. Nakagawa, M. Tamura and K. Tomishige, J. Mater. Chem. A, 2014, 2, 6688 CAS.
- J. Deutsch, A. Martin and H. Lieske, J. Catal., 2007, 245, 428 CrossRef CAS PubMed.
- C. Wei, C. Dasari and M. A. Suppes, AIChE J., 2006, 52, 3543 CrossRef PubMed.
- S. K. Tyrlik, D. Szerszen, M. Olejnik and W. Danikiewicz, J. Mol. Catal. A: Chem., 1996, 106, 223 CrossRef CAS.
- T. Miyazawa, Y. Kusunoki, K. Kunimori and K. Tomishige, J. Catal., 2006, 240, 213 CrossRef CAS PubMed.
- J. T. Dam and U. Hanefeld, ChemSusChem, 2011, 4, 1017 CrossRef PubMed.
- C. Montassier, D. Giraud, J. Barbier and J. P. Boitiaux, Bull. Soc. Chim. Fr., 1989, 2, 148 Search PubMed.
- C. Montassier, J. C. Menezo, J. Moukol and J. Naja, J. Mol. Catal., 1991, 70, 65 CrossRef CAS.
- S. Wang, K. Yin, Y. Zhang and H. Liu, ACS Catal., 2013, 3, 2112 CrossRef CAS.
- Y. Shinmi, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2010, 94, 318 CrossRef CAS PubMed.
- Y. Nakagawa, Y. Shinmi, S. Koso and K. Tomishige, J. Catal., 2010, 272, 191 CrossRef CAS PubMed.
- Y. Amada, Y. Shinmi, S. Koso, T. Kubota, Y. Nakagawa and K. Tomishige, Appl. Catal., B, 2011, 205, 117 CrossRef PubMed.
- D. Durán-Martín, M. Ojeda, M. López Granados, J. L. G. Fierro and R. Mariscal, Catal. Today, 2013, 210, 98 CrossRef PubMed.
- M. A. Dasari, P.-P. Kiatsimkul, W. R. Sutterlin and G. J. Suppes, Appl. Catal., A, 2005, 281, 225 CrossRef CAS PubMed.
- R. B. Mane, A. A. Ghalwadkar, A. M. Hengne, Y. R. Suryawanshi and C. V. Rode, Catal. Today, 2011, 164, 447 CrossRef CAS PubMed.
- N. D. Kim, J. R. Park, D. S. Park, B. K. Kwak and J. H. Yi, Green Chem., 2012, 14, 2638 RSC.
- Z. W. Huang, F. Cui, H. X. Kang, J. Chen, X. Z. Zhang and C. G. Xia, Chem. Mater., 2008, 20, 5090 CrossRef CAS.
- Z. W. Huang, F. Cui, H. X. Kang, J. Chen and C. G. Xia, Appl. Catal., A, 2009, 366, 288 CrossRef CAS PubMed.
- Z. W. Huang, F. Cui, J. J. Xue, J. L. Zuo, J. Chen and C. G. Xia, Catal. Today, 2012, 190, 37 Search PubMed.
- R. B. Mane, A. M. Hengne, A. A. Ghalwadkar, S. Vijayanand, P. H. Mohite, H. S. Potdar and C. V. Rode, Catal. Lett., 2010, 135, 141 CrossRef CAS.
- R. B. Mane, S. E. Kondawar, P. S. Niphadkar, P. N. Joshi, K. R. Patil and C. V. Rode, Catal. Today, 2012, 198, 321 CrossRef CAS PubMed.
- A. Wolosiak-Hnat, E. Milchert and B. Grzmil, Chem. Eng. Technol., 2013, 36, 411 CrossRef CAS PubMed.
- M. Balaraju, K. Jagadeeswaraiah, P. S. Sai Prasad and N. Lingaiah, Catal. Sci. Technol., 2012, 2, 1967 CAS.
- Z. L. Yuan, J. H. Wang, L. N. Wang, W. H. Xie, P. Chen, Z. Y. Hou and X. M. Zheng, Bioresour. Technol., 2010, 101, 7088 CrossRef CAS PubMed.
- M. Balaraju, V. Rekha, P. S. Sai Prasad, R. B. N. Prasad and N. Lingaiah, Catal. Lett., 2008, 126, 119 CrossRef CAS.
- C. V. Rode, R. B. Mane, A. S. Potdar, P. B. Patil, P. S. Niphadkar and P. N. Joshi, Catal. Today, 2012, 190, 37 CrossRef PubMed.
- S. X. Xia, R. F. Nie, X. Y. Lu, L. N. Wang, P. Chen and Z. Y. Hou, J. Catal., 2012, 296, 1 CrossRef CAS PubMed.
- S. X. Xia, Z. L. Yuan, L. N. Wang, P. Chen and Z. Y. Hou, Appl. Catal., A, 2011, 403, 173 CrossRef CAS PubMed.
- E. S. Vasiliadou and A. A. Lemonidou, Appl. Catal., A, 2011, 396, 177 CrossRef CAS PubMed.
- E. S. Vasiliadou, T. M. Eggenhuisen, P. Munnik, P. E. de Jongh, K. P. de Jong and A. A. Lemonidou, Appl. Catal., B, 2014, 145, 108 CrossRef CAS PubMed.
- T. Sánchez, P. Salagre, Y. Cesteros and A. Bueno-López, Chem. Eng. J., 2012, 179, 302 CrossRef PubMed.
- S. H. Zhu, X. Q. Gao, Y. L. Zhu, Y. F. Zhu, H. Y. Zheng and Y. W. Li, J. Catal., 2013, 303, 70 CrossRef CAS PubMed.
- L. Y. Guo, J. X. Zhou, J. B. Mao, X. W. Guo and S. G. Zhang, Appl. Catal., A, 2009, 367, 93 CrossRef CAS PubMed.
- H. C. Liu, Catal. Lett., 2007, 117, 62 CrossRef.
- J. X. Zhou, L. Y. Guo, X. W. Guo, J. B. Mao and S. G. Zhang, Green Chem., 2010, 12, 1835 RSC.
- C.-J. Yue, L.-P. Gu, Y. Su and S.-P. Zhu, React. Kinet., Mech. Catal., 2014, 111, 633 CrossRef CAS.
- Z. H. Xiao, X. K. Wang, J. H. Xiu, Y. M. Wang, C. T. Williams and C. H. Liang, Catal. Today, 2014, 234, 200 CrossRef CAS PubMed.
- Z. Q. Ma, Z. H. Xiao, J. A. van Bokhoven and C. H. Liang, J. Mater. Chem., 2010, 20, 755 RSC.
- Z. H. Xiao, J. H. Xiu, X. K. Wang, B. S. Zhang, C. T. Williams, D. S. Su and C. H. Liang, Catal. Sci. Technol., 2013, 3, 1108 CAS.
- C. H. Liang, Z. Q. Ma, L. Ding and J. S. Qiu, Catal. Lett., 2009, 130, 169 CrossRef CAS.
- Z. H. Xiao, S. H. Jin, X. K. Wang, W. Z. Li, J. H. Wang and C. H. Liang, J. Mater. Chem., 2012, 22, 16598 RSC.
- Z. J. Wu, Y. Z. Mao, M. Song, X. Q. Yin and M. H. Zhang, Catal. Commun., 2013, 32, 52 CrossRef CAS PubMed.
- B. K. Kwak, D. S. Park, Y. S. Yun and J. Yi, Catal. Commun., 2012, 24, 90 CrossRef CAS PubMed.
- H. Tan, M. N. Hedhill, Y. L. Wang, J. Z. Zhang, K. Li, S. Sioud, Z. A. Al-Talla, M. H. Amad, T. Zhan, O. E. Tall and Y. Han, Catal. Sci. Technol., 2013, 3, 3360 CAS.
- A. Perosa and P. Tundo, Ind. Eng. Chem. Res., 2005, 44, 8535 CrossRef CAS.
- W. Q. Yu, J. Xu, H. Ma, C. Chen, J. Zhao, H. Miao and Q. Song, Catal. Commun., 2010, 11, 493 CrossRef CAS PubMed.
- A. Marinoiu, G. Ionita, C.-L. Gaspar, C. Cobzaru and S. Oprea, React. Kinet. Catal. Lett., 2009, 97, 315 CrossRef CAS.
- E. V. Ryneveld, A. S. Mahomed, P. S. van Heerden, M. J. Green and H. B. Friedrich, Green Chem., 2011, 13, 1819 RSC.
- J. H. Huang and J. X. Chen, Chin. J. Catal., 2012, 33, 790 CrossRef CAS.
- I. Gandarias, P. L. Arias, J. Requies, M. E. Doukkali and M. B. Güemez, J. Catal., 2011, 282, 237 CrossRef CAS PubMed.
- A. Marinoiu, G. Ionita, C.-L. Gaspar, C. Cobzaru, D. Marinescu, C. Teodorescu and S. Oprea, React. Kinet., Mech. Catal., 2010, 99, 111 CAS.
- X. H. Guo, Y. Li, Q. Y. Liu and W. J. Shen, Chin. J. Catal., 2012, 33, 645 CrossRef CAS.
- X. Y. Guo, A. Y. Yin, X. D. Guo, X. Y. Guo, W. L. Dai and K. N. Fan, Chin. J. Chem., 2011, 29, 1563 CrossRef CAS PubMed.
- X. H. Guo, Y. Li, W. Song and W. J. Shen, Catal. Lett., 2011, 141, 1458 CrossRef CAS.
- Y. B. Cao, X. Zhang, J. M. Fan, P. Hu, L.-Y. Bai, H.-B. Zhang, F.-L. Yuan and Y.-F. Chen, Cryst. Growth Des., 2011, 11, 2 Search PubMed.
- M. Checa, F. Auneau, J. Hidalgo-Carrillo, A. Marinas, J. M. Marinas, C. Pinel and F. J. Urbano, Catal. Today, 2012, 196, 91 CrossRef CAS PubMed.
- I. Gandarias, P. L. Arias, J. Requies, M. B. Güemez and J. L. G. Fierro, Appl. Catal., B, 2010, 97, 248 CrossRef CAS PubMed.
- Z. L. Yuan, P. Wu, J. Gao, X. Y. Lu, Z. Y. Hou and X. M. Zheng, Catal. Lett., 2009, 130, 261 CrossRef CAS.
- R. Rodrigues, N. Isoda, M. Gonçalves, F. C. A. Figueiredo, D. Mandelli and W. A. Carvalho, Chem. Eng. J., 2012, 198, 457 CrossRef PubMed.
- M. L. Barbelli, G. F. Santori and N. N. Nichio, Bioresour. Technol., 2012, 111, 500 CrossRef CAS PubMed.
- C. H. Deng, X. Z. Duan, J. H. Zhou, D. Chen, X. G. Zhou and W. K. Yuan, Catal. Today, 2014, 234, 208 CrossRef CAS PubMed.
- L. J. Liu, Y. H. Zhang, A. Q. Wang and T. Zhang, Chin. J. Catal., 2012, 33, 1257 CrossRef CAS.
- L. F. Gong, Y. Lu, Y. J. Ding, R. H. Lin, J. W. Li, W. D. Dong, T. Wang and W. M. Chen, Appl. Catal., A, 2010, 390, 119 CrossRef CAS PubMed.
- Y. H. Zhang, X.-C. Zhao, Y. Wang, L. K. Zhou, J. Y. Zhang, J. Wang, A. Q. Wang and T. Zhang, J. Mater. Chem. A, 2013, 1, 3724 CAS.
- S. H. Zhu, Y. N. Qiu, Y. L. Zhu, S. L. Hao, H. Y. Zheng and Y. W. Li, Catal. Today, 2013, 212, 120 CrossRef CAS PubMed.
- R. Arundhathi, T. Mizugaki, T. Mitsudome, K. Jitsukawa and K. Kaneda, ChemSusChem, 2013, 6, 1345 CrossRef CAS PubMed.
- Y. Nakagawa, X. H. Ning, Y. Amada and K. Tomishige, Appl. Catal., A, 2012, 433, 128 CrossRef PubMed.
- E. G. Suarez, M. P. Cadenas, A. G. Ruiz, I. R. Ramos and A. Arcoya, Appl. Surf. Sci., 2013, 287, 108 CrossRef PubMed.
- T. Miyazawa, S. Koso, K. Kunimori and K. Tomishige, Appl. Catal., A, 2007, 329, 30 CrossRef CAS PubMed.
- P. Centomo, V. Nese, S. Sterchele and M. Zecca, Top. Catal., 2013, 56, 822 CrossRef CAS.
- E. S. Vasiliadou, E. Heracleous, I. A. Vasalos and A. A. Lemonidou, Appl. Catal., B, 2009, 92, 90 CrossRef CAS PubMed.
- E. S. Vasiliadou and A. A. Lemonidou, Org. Process Res. Dev., 2011, 15, 925 CrossRef CAS.
- J. Feng, W. Xiong, B. Xu, W. D. Jiang, J. B. Wang and H. Chen, Catal. Commun., 2014, 46, 98 CrossRef CAS PubMed.
- S.-H. Lee and D. J. Moon, Catal. Today, 2011, 174, 10 CrossRef CAS PubMed.
- T. Jiang, Y. X. Zhou, S. G. Liang, H. Z. Liu and B. X. Han, Green Chem., 2009, 11, 1000 RSC.
- H. Z. Liu, S. G. Liang, T. Jiang, B. X. Han and Y. X. Zhou, Clean: Soil, Air, Water, 2012, 40, 318 CrossRef CAS PubMed.
- L. Ma and D. H. He, Top. Catal., 2009, 52, 834 CrossRef CAS.
- R. P. Wang, X. W. Guo, X. S. Wang, J. Q. Hao, G. Li and J. H. Xiu, Appl. Catal., A, 2004, 261, 7 CrossRef CAS PubMed.
- R. P. Wang, X. W. Guo, X. S. Wang and J. Q. Hao, Catal. Today, 2004, 93–95, 217 CrossRef CAS PubMed.
- X. W. Guo, R. P. Wang, X. S. Wang and J. Q. Hao, Catal. Today, 2004, 93–95, 211 CrossRef CAS PubMed.
- J. X. Wang, M. Liu, X. W. Guo, H. Y. Wu, J. Xu and W. Q. Lu, React. Kinet., Mech. Catal., 2011, 102, 447 CrossRef CAS.
- C. F. Wang, X. W. Guo, X. S. Wang, R. P. Wang and J. Q. Hao, Catal. Lett., 2004, 96, 79 CrossRef CAS.
- G. D. Yadav, P. A. Chandan and D. P. Tekale, Ind. Eng. Chem. Res., 2012, 51, 1549 CrossRef CAS.
- J. X. Zhou, J. Zhang, X. W. Guo, J. B. Mao and S. G. Zhang, Green Chem., 2012, 14, 156 RSC.
- I. Gandarias, S. G. Fernandez, M. L. Doukkali, J. Requies and P. L. Arias, Top. Catal., 2013, 56, 995 CrossRef CAS.
- R. A. W. Johnstone, A. H. Wilby and I. D. Entwistle, Chem. Rev., 1985, 85, 129 CrossRef CAS.
- A. Wolfson, C. Dlugy, Y. Shotland and D. Tavor, Tetrahedron Lett., 2009, 50, 5951 CrossRef CAS PubMed.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964 CrossRef CAS PubMed.
- E. D′Hont, S. van de Vyver, B. F. Sels and P. A. Jacobs, Chem. Commun., 2008, 6011 RSC.
- D. Roy, B. Subramaniam and R. V. Chaudhari, Catal. Today, 2010, 156, 31 CrossRef CAS PubMed.
- I. Gandarias, P. L. Arias, S. G. Fernández, J. Requies, M. E. Doukkali and M. B. Güemez, Catal. Today, 2012, 195, 22 CrossRef CAS PubMed.
- I. Gandarias, P. L. Arias, J. Requies, M. E. Doukkali and M. B. Güemez, J. Catal., 2011, 282, 237 CrossRef CAS PubMed.
- M. G. Musolino, L. A. Scarpino, F. Mauriello and R. Pietropaolo, Green Chem., 2009, 11, 1511 RSC.
- I. Gandarias, J. Requies, P. L. Arias, U. Armbruster and A. Martin, J. Catal., 2012, 290, 79 CrossRef CAS PubMed.
- J. Yuan, S. H. Li, L. Yu, Y. M. Liu and Y. Cao, Chin. J. Catal., 2013, 34, 2066 CrossRef CAS.
- T. Miyazawa, S. Koso, K. Kunimori and K. Tomishige, Appl. Catal., A, 2007, 318, 244 CrossRef CAS PubMed.
- S. X. Xia, Z. L. Yuan, L. N. Wang, P. Chen and Z. Y. Hou, Bioresour. Technol., 2012, 104, 814 CrossRef CAS PubMed.
- A. Bienholz, F. Schwab and P. Claus, Green Chem., 2010, 12, 290 RSC.
- A. Wołosiak-Hnat, E. Milchert and G. Lewandowski, Org. Process Res. Dev., 2013, 17, 701 CrossRef.
- C. V. Rode, A. A. Ghalwadkar, R. B. Mane, A. M. Hengne, S. T. Jadkar and N. S. Biradar, Org. Process Res. Dev., 2010, 14, 1385 CrossRef CAS.
- L. Ma, D. He and Z. Li, Catal. Commun., 2008, 9, 2489 CrossRef CAS PubMed.
- D. G. Lahr and B. H. Shanks, J. Catal., 2005, 232, 386 CrossRef CAS PubMed.
- Y. H. Feng, H. B. Yin, A. L. Wang, L. Q. Shen, L. B. Yu and T. S. Jiang, Chem. Eng. J., 2011, 168, 403 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |