DOI:
10.1039/C5RA08551A
(Paper)
RSC Adv., 2015,
5, 74605-74610
Identification of disulfide isomerase ERp57 as a target for small molecule cardioprotective agents†
Received
8th May 2015
, Accepted 14th August 2015
First published on 14th August 2015
Abstract
We previously reported a novel danshensu analogue known as ADTM, which exhibited strong protective effects against oxidative stress-induced cellular injury and acute ischemic myocardial infarct in rat; however, the exact protein target of ADTM has not been fully characterized. In the present study, a biotin-conjugated ADTM analogue (BAA) was employed as molecular probe to identify its protein targets. BAA exhibited similar protective effect against oxidative stress-induced cell injury in H9c2 cardiomyoblast. A chemical proteomic approach identified ERp57 as the specific target for BAA. Further evaluation with Western blot and immunofluorescence staining assays confirmed the direct interactions between BAA and ERp57. Moreover, BAA displayed potent inhibitory effect on the catalytic activity of ERp57 in the insulin reduction assay. Molecular docking showed that BAA bound at the active site of ERp57. These data suggested that ERp57 is a potential target of cardioprotective danshensu analogues, and provided the basis for the further optimization of the cardioprotective compounds.
1. Introduction
The search for safe and effective substances is considered to be the “holy grail” in modern pharmaceutical discovery. The vast majority of the chemical drugs are small molecules with remarkably simple structures, but they usually induce complex pharmacological effects in vivo. It is a major challenge to identify the druggable targets with specificity of these molecules with their intertwined multiple pharmacological actions when the chemical drugs enter a world of chemical reactions in the human body. It has been reported that there are at least 600–1500 proteins that could serve as drug targets for small molecules,1,2 but in 2011, only 435 proteins in the human genome were reported to be the drug targets for 989 drugs via 2242 drug-target interactions.3 This indicates that there are a number of potential novel drug targets remaining to be explored for drug discovery.
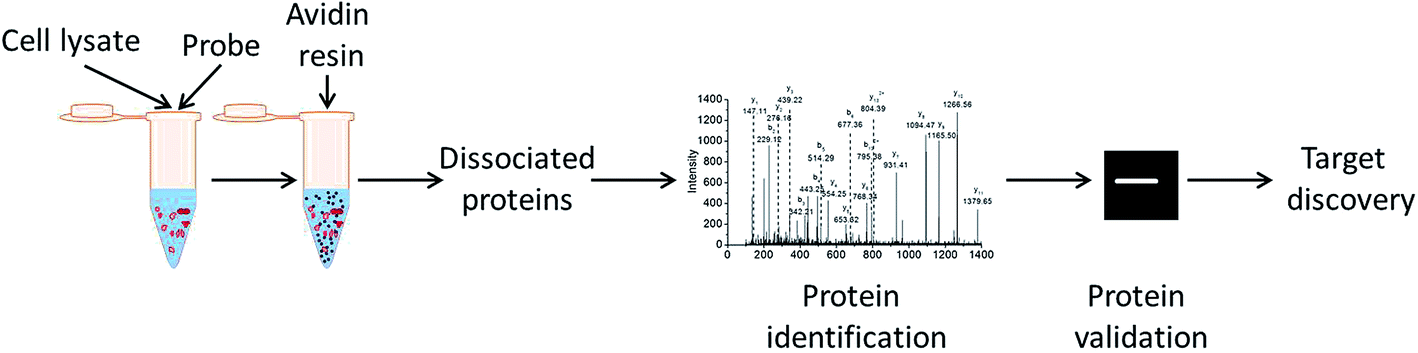
Molecular target identification is one of the most important facets for drug discovery. From 1999 to 2013, 78% of the first-in-class drugs approved by the US Food and Drug Administration were discovered based on target-based approaches.4 Among various methods for drug target identification described to date, chemical proteomics is one of the most powerful method for identifying the target of small molecule.5 In general, solid phase carrier such as resin beads or Microtiter plates conjugated with compounds with suitable functional groups (e.g. biotin) is used to pull-down and isolate the binding proteins. The proteins bound to the solid carrier is eluted, and identified by mass spectrometry (MS) allowing rapid identification of the profiles of the bound-proteins.
Protein disulfide isomerase family (PDI) member ERp57 has received much attention as a potential druggable target in recent years. ERp57 is observed to be related to various cellular functions including apoptosis and platelet activity. For instance, it was reported that pharmacologically inhibiting ERp57 activity eliminated the effects of various apoptotic inducers.6,7 In addition, the knockdown of ERp57 by RNA interference enhanced the apoptosis mediated by fenretinide (an endoplasmic reticulum inducer) in cancer cells.8 ERp57 knockout mice study also showed that ERp57 down-regulation could contribute to the increase in lifespan in mammals.9 On the other hand, mice which were ERp57-deficient or treated with anti-ERp57 antibody showed that inhibition of ERp57 correlated to the suppression of platelet aggregation and thrombus formation in vivo.10,11 Separate lines of evidences point towards the pharmacological inhibition of ERp57 as potential strategy for disease therapy and/or for health maintenance. However, specific chemical inhibitor for ERp57 is not yet available.
We previously reported a novel compound named ADTM with strong cardioprotective properties.12 ADTM was synthesized in our laboratory by conjugating two well-known cardiovascular protective compounds, danshensu and tetramethylpyrazine, which are present in Chinese medicinal plants, danshen (Salvia miltiorrhiza) and chuanxiong (Ligustium wallichii Franch).12 ADTM exhibited strong cardioprotective effects against oxidative stress-induced cellular injury and acute ischemic myocardial infarct in rat.13 In this study, biotinylated-ADTM analogue (BAA) was employed as small molecular probe for the identification of the protein targets of ADTM. By the approach of chemical proteomics through liquid chromatography with tandem mass spectrometry (LC-MS/MS), the profile of the pull-down proteins bound to BAA were analyzed. The results identified ERp57 as a specific target of ADTM, providing a promising opportunity for developing novel cardioprotective agents with selective activity at ERp57 by using BAA as a template.
2. Materials and methods
2.1 Chemicals
ADTM and BAA (purity > 95%) were synthesized at Jinan University, China. Human recombinant ERp57 was obtained from Abcam (Cambridge, UK). MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide], insulin, DMSO (dimethyl sulfoxide), DTT (dithiothreitol) and tert-butylhydroperoxide (t-BHP) were bought from Sigma-Aldrich (St. Louis, MO). DMEM, fetal bovine serum (FBS), penicillin and streptomycin were purchased from Gibco Life Technologies (Grand Island, USA).
2.2 Cell culture
The H9c2 cell line was purchased from the American Tissue Type Collection (Manassas, USA). Cells were cultured in complete medium containing DMEM supplemented with 10% FBS, 100 U ml−1 of penicillin and 100 U ml−1 of streptomycin in 75 cm2 tissue culture flasks, and cultured at 37 °C, in a humidified incubator containing 5% CO2. The cells were fed every 2 days, and sub-cultured once they reached 70–80% confluence.
2.3 Cell treatment and MTT assay
To measure cell viability, cells were seeded in 96-well plates at a density of 5 × 103 cells per well. The cultures were grown at 37 °C for 24 h (hours). After discarding the old medium, the cells were pretreated for 2 h with varying concentrations of ADTM or BAA diluted in DMEM nutrient mixture containing 0.5% FBS, followed by treatment with 150 μM t-BHP for 4 h, and cell viability was measured by the MTT assay. Cells incubated with 0.1% DMSO served as controls.
2.4 Protein preparation from H9c2 cells
H9c2 cells were lysed using a dounce homogenizer in NP-40 lysis buffer (Beyotime, China) with 1 mM phenylmethylsulfonyl fluoride (PMSF). Plate lysates were centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g for 10 min at 4 °C, and the supernatant was collected and stored at −80 °C until further use.
500 g for 10 min at 4 °C, and the supernatant was collected and stored at −80 °C until further use.
2.5 Streptavidin agarose beads pull-down with BAA
The H9c2 cells lysates (3 μg μl−1) were exposed to streptavidin agarose beads (Pierce Biotech, Rockford, IL, USA) for 2 h at 4 °C and then centrifuged for 3 min at 2500 g. The beads were discarded to remove endogenous biotin. Aliquots of supernatants (300 μl) were treated with BAA (300 μM) for 2 h. The supernatant was combined with beads and allowed to shake overnight at 4 °C. To eliminate the nonspecific combination with compound or beads before protein analysis, 0.1% DMSO and biotin were added to one aliquot of lysate as negative controls. The compound-protein bound to beads was digested with trypsin in 37 °C overnight and each injected to LC-MS. Sample 1 (DMSO) and sample 2 (biotin) being the negative control and sample 3 being rich protein group.
2.6 Nano LC-MS/MS analysis and data analysis
Nano-flow LC separation experiments were performed using Agilent 1200 series nano pump. Solvent A (2% ACN, 0.5% formic acid in water) and B (98% ACN, 0.5% formic acid in water) were used to provide pH 2 organic gradient. Reserve phase C18 separation with 60 min 5–35% solvent B gradient was applied for all three samples. TripleTOF 5600 system (AB SCIEX, Concord, Canada) fitted with a Nanospray III source (AB SCIEX, Concord, Canada); the parameters used were as follows: ion spray voltage, 2.8 kV; curtain gas, 30 psi; nebulizer gas, 6 psi; interface heater temperature, 150 °C. For IDA, full scans were acquired within 250 ms over the range m/z 400–1250, followed by MS/MS scans of the 20 most abundant peaks that exceeded 125 counts per second and carried a charge between +2 to +5 in the range m/z 100–1500. The dynamic exclusion time of the acquired ions was set at 20 s. The acquired MS/MS data were analyzed using the Paragon algorithm in ProteinPilot 4.0 software (Applied Biosystems, Framingham, USA).14 They were searched against theoretical spectra generated from sequences in Uniprot (http://www.uniprot.org). Uniprot Rat complete proteome database released in August 2011 (34098 entries) was used.
2.7 Insulin reduction assay
ERp57 activity was measured with an assay that measures the catalytic reduction of insulin, as described previously with a minor modification.15,16 Briefly, insulin (1 mg ml−1) was prepared in assay buffer (100 mM potassium phosphate, 1 mM EDTA and 8 μM DTT). 50 μl of insulin was incubated in the presence of 10 μl of enzyme and varying concentrations of BAA, and then 10 μl of 1 mM DTT was added. The 96-well plate was incubated at 25 °C, and then the absorbance at 630 nm was read on a microplate reader (Perkin-Elmer, Singapore) for each sample consecutively at 4 min intervals for 40 min. ERp57 enzyme inhibition in the presence of compound was determined by the following formula: enzyme inhibition (%) = [1 − (OD[compound+ERp57+DTT] − OD[DTT])/(OD[ERp57+DTT] − OD[DTT])] × 100%.
2.8 Western blot analysis
For Western blot validation study, beads bound proteins were washed four times in 0.3 ml lysis buffer and remaining complexes were resuspended in 2 × SDS sample loading buffer, followed by a 5 min boil. The supernatant was analyzed by Western blotting as previously described method.13 Briefly, after incubation with anti-ERp57 antibody (Santa Cruz Biotechnology, Santa Cruz, CA), the membrane was incubated for 1 h with secondary horseradish peroxidase-conjugated antibody (Cell Signaling Technology, Beverly, MA). Proteins were detected by an advanced enhanced ECL system (GE Healthcare, Little Chalfont, UK).
2.9 Immunofluorescence staining
H9c2 cells were incubated with 100 μM BAA for 1 h, then washed three times with ice-cold PBS, and were fixed by 4% paraformaldehyde for 15 min at room temperature. The cells were then washed three times with PBS containing 1% BSA, and were incubated with blocking buffer containing 0.3% TritonX-100 and 1% BSA in PBS for 45 min at room temperature. The cells were incubated with anti-ERp57 antibody (1:250) overnight at 4 °C. The cells were then washed three times with PBS, and were incubated with Alexa Fluor 680-conjugated antibody (1:200) (Invitrogen, Eugene, OR, USA) and Streptavidin-FITC (1:500) for 45 min at room temperature. To visualize the nuclear, the cells were counterstained with 300 nM DAPI for 10 min. Fluorescence imaging of the cells was performed with IN Cell Analyzer 2000 (GE Healthcare UK Ltd, Buckinghamshire, UK).
2.10 Molecular docking study
Molecular docking of BAA to ERp57 was performed using the three dimensional crystal structure of human ERp57 (PDB: 3F8U) obtained from the Protein Data Bank. The software AutoDock 4.2 was used for all dockings in this study as previously described method.17 In general, the docking parameters for AutoDock were kept to their default values. The 3D structures of BAA were converted using Open Babel software, followed by energy minimization with MMFF94s force field along with addition of hydrogen atoms.18 The grid box was 42.2 Å × 56.6 Å × 79.2 Å, encompassing the active site cavity (CYS409) of human ERp57. Top 10 configurations of binding energy were ranked by AutoDock, and the lowest energy configuration was selected. The binding results were illustrated as three dimensional (3-D) by PyMOL Molecular Graphics System Version 1.3 (Schrödinger, LLC, New York, USA).
2.11 Statistical analysis
The MTT assay was performed three times. Data are presented as mean ± SD. The groups were compared using one-way ANOVA followed by Tukey's multiple comparison tests using the statistics module of Graph Pad Prism 5.0. A value of P < 0.05 was considered statistically significant.
3. Results and discussion
3.1 Protective effects of BAA against t-BHP-induced cell injury in H9c2 cells
We have previously reported that ADTM showed prominent anti-oxidative effects in reducing t-BHP-induced cell injury in H9c2 cells.13 t-BHP is a hydrogen peroxide donor is often used to mimic oxidative stress in myocardial ischemia.19 Here, we examined the cardioprotective actions of the biotin-conjugated ADTM analogue (BAA) by using t-BHP-induced cell injury in H9c2 cells as a cellular model of cardiac injury. Results in Fig. 1B showed that the pretreatment of BAA (3, 10, 30 and 100 μM) greatly attenuated t-BHP-induced cell injury in a concentration-dependent manner. The protective efficiency of BAA was comparable to that of ADTM and was therefore considered to be an eligible tool for the identification of potential protein target of ADTM in our recently published work.25
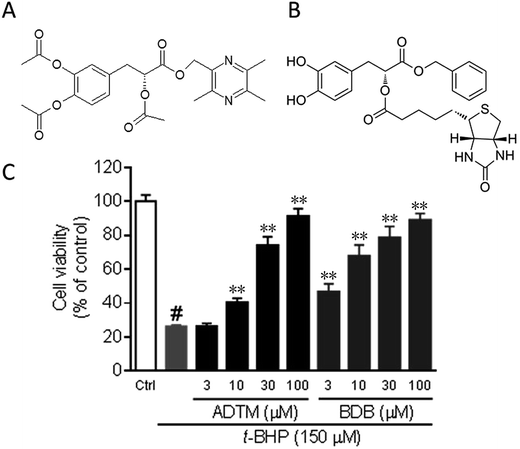 |
| | Fig. 1 Evaluation of protective effect of ADTM and BAA on t-BHP-induced cell injury in H9c2 cells. (A) Chemical structures of ADTM and BAA. (B) Cell viability of H9c2 cells incubated with different concentrations (3, 10, 30, 100 μM) of ADTM or BAA for 4 h. Pretreatment with ADTM or BAA significantly attenuated t-BHP-induced cell injury. #P < 0.05 versus control group, **P < 0.05 versus t-BHP treatment group. | |
 |
| | Fig. 2 Schematic illustration of identifying BAA-protein interactions with chemical proteomics. | |
 |
| | Fig. 3 Identification of ERp57 as one target of BAA in H9c2 proteins. ERp57 was identified by chemical proteomic analysis. | |
3.2 Targets profiling of BAA by chemical proteomics approach
To isolate the protein targets that bound to BAA, H9c2 cell lysates were incubated with BAA and the BAA-protein complexes were enriched with streptavidin agarose beads followed by protein profiling using LC-MS/MS (Fig. 2 and 3). DMSO and biotin served as negative controls to eliminate protein bound non-specifically. Eight proteins were identified with >95% protein identification probability (ESI,† Table 1). In particular, three of the identified proteins were glycoprotein-specific members of the PDI family, ERp72, ERp57 and ERp5, which are well known to exhibit disulfide bonds modifying activity and subsequently correct misfolded proteins to achieve homeostasis.20 In another set of experiment performed with rat blood platelet lysates, BAA-bound proteins pulled down were also identified as ERp72, ERp57 and ERp5 (data not shown). These results indicated that ERp72, ERp57 and ERp5 are protein targets of BAA independent of the protein tissues. In addition to the PDI protein family members, peroxiredoxin-1, peroxiredoxin-2, 14-3-3 protein zeta/delta, and annexin A1 were also found to bind to BAA. Peroxiredoxin-1 and peroxiredoxin-2 belong to the antioxidant enzyme family peroxiredoxin. 14-3-3 protein zeta/delta and annexin A1, which are proteins involved in the regulation of phospholipase A2 activity and calcium binding respectively, are also known to be implicated in platelet activation and heart failure.21,22 The final identified protein was translationally-controlled tumor protein, which was found to be involved in calcium binding and microtubule stabilization.23
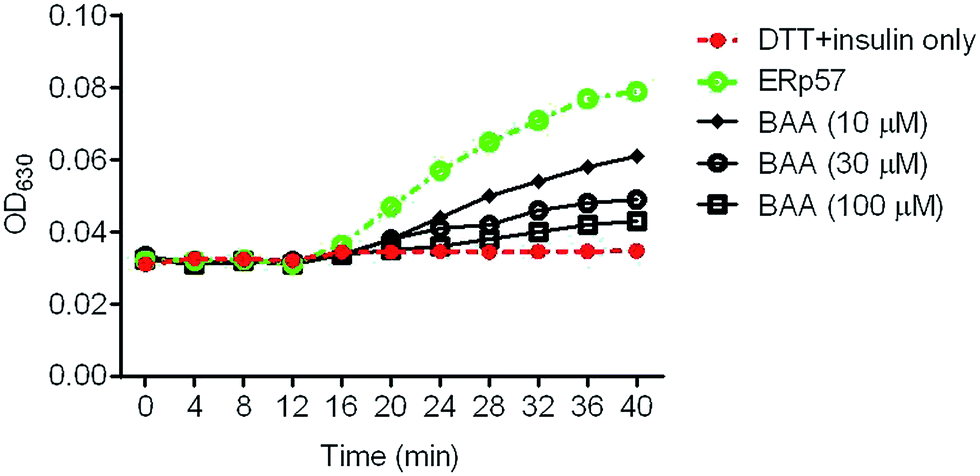
3.3 BAA inhibited the redox activity of ERp57
To investigate whether BAA could directly modulate the enzymatic activity of ERp57, the effect of BAA on ERp57 activity was evaluated by insulin reduction assay. The result showed that BAA inhibited the activity of ERp57 in a concentration- and time-dependent manner (Fig. 4). The half maximal inhibitory concentration (IC50) of BAA on ERp57 activity was about 10–30 μM. In line with these results, ERp57 has recently been implicated to play crucial roles in oxidative stress-induced apoptotic cell death by hyperoxia and tunicamycin.7 It is likely that ERp57 acts as a target for oxidative stress induced by hydrogen peroxide.24 However, further studies are needed to confirm this notion.
 |
| | Fig. 4 BAA inhibits the enzymatic activity of ERp57 in a concentration- and time-dependent manner. The recombinant ERp57 was incubated with different concentrations of BAA for 40 min, and its catalytic activity was determined in insulin reduction assay. | |
3.4 Evaluation of the binding between BAA and ERp57 by Western blot analysis and immunofluorescence staining
The binding between BAA and ERp57 was further evaluated by Western blot analysis by using anti-ERp57 antibody in H9c2 cell lysates incubated with BAA. As shown in Fig. 5A, specific anti-ERp57 antibody yielded a clear single band with a molecular mass of approximately 61 kDa with BAA but not DMSO or biotin. Furthermore, in our recently published paper, we found that ADTM (unbiotinlyated compound) competitively inhibited the binding of BAA with ERp57, providing further evidence that ADTM was a reversible ligand at ERp57.25 In double immunofluorescence staining assay, anti-ERp57 antibody (red) and Streptavidin-FITC against BAA (green) showed that BAA was co-localized with ERp57 protein in cytoplasm of H9c2 cells (Fig. 5B). This result provided further evidence of the direct interaction between BAA and ERp57 in cellular environment.
 |
| | Fig. 5 Target validation of ERp57 in H9c2 cells. (A) Confirmation of BAA targeted protein isolated from H9c2 cardiomyoblast cells by Western blot. (B) BAA colocalized with ERp57 in H9c2 cells. After treatment with or without 100 μM BAA for 2 h, H9c2 cells were stained with ERp57 antibody (red), Streptavidin-FITC (green) for biotin and DAPI (blue) for nuclear. Fluorescence imaging of the cells was performed automatically with IN Cell Analyzer 2000 (at 60× magnification). | |
3.5 Molecular docking analysis
The protein structure of ERp57 has been reported to contain two catalytically active CYS-GLY-HIS-CYS motifs (CYS57-GLY58-HIS59-CYS60 and CYS406-GLY407-HIS408-CYS409), with CYS409 as the major active site for pharmacological function.10,26 Therefore, ERp57 protein domain (PDB: 3F8U) enclosing the catalytic center (CYS409) was chosen for the molecular docking analysis between BAA and ERp57. BAA bound to ERp57 with a binding free energy of −9.21 kcal mol−1 (Fig. 6), forming hydrogen bonds with the side chains of GLY178, PRO179, TRP221, ALA219, ALA217, CLY407, HIS408 and PHE450 in the catalytic center (CYS409) of ERp57.
 |
| | Fig. 6 Molecular docking simulation of the interaction between BAA and ERp57. The interaction between BAA and ERp57 was viewed by the representative structure (best energy) of the energetically best structure clusters found by AutoDock. The free binding energy to the active cavity of ERp57 (PDB: 3F8U) is −9.22 kcal mol−1. The yellow stick represents BAA and the crystal structure of ERp57 domain with each monomer presented in ribbon diagram. | |
4. Conclusion
Taken together, our present study reported the biotin-conjugated ADTM analogue (BAA) displayed protective effects with similar efficacy as ADTM. BAA protected H9c2 cells from oxidative stress-induced cell injury. BAA displayed specific binding at ERp57 and inhibited its catalytic activity. Molecular docking showed that BAA displayed direct binding at ERp57 and it would be interesting to define the binding confirmation of the BAA-ERp57 complex by X-ray co-crystallography or NMR spectroscopy experiments in the future. Our data provided a rationale for the further development of cardioprotective agent targeting ERp57 against oxidative stress-induced cardiac injury.
Acknowledgements
This work is supported by the research fundings from the University of Macau (Project no. SRG2014-00025, MYRG138 (Y1-Y4)-ICMS12-LMY, MYRG2015-00214-ICMS-QRCM and MYRG2015-00161-ICMS-QRCM), the National Natural Science Foundation of China (Grant No. 81460552, 81403139 and 81302776), the Overseas and Hong Kong, Macau Young Scholars Collaborative Research Fund by the Natural National Science Foundation of China (Grant No. 81328025), the Research Fund of State Key Laboratory of Chinese Medicine and Molecular Pharmacology (Incubation), Shenzhen, and Science and Technology Development Fund of Macau SAR (Grant No. 078/2011/A3 and 014/2011/A1).
Notes and references
- A. P. Russ and S. Lampel, Drug Discovery Today, 2005, 10, 1607–1610 CrossRef.
- A. L. Hopkins and C. R. Groom, Nat. Rev. Drug Discovery, 2002, 1, 727–730 CrossRef CAS PubMed.
- M. Rask-Andersen, M. S. Almen and H. B. Schioth, Nat. Rev. Drug Discovery, 2011, 10, 579–590 CrossRef CAS PubMed.
- J. Eder, R. Sedrani and C. Wiesmann, Nat. Rev. Drug Discovery, 2014, 13, 577–587 CrossRef CAS PubMed.
- K. Wierzba, M. Muroi and H. Osada, Curr. Opin. Chem. Biol., 2011, 15, 57–65 CrossRef CAS PubMed.
- G. Zhao, H. Lu and C. Li, J. Biol. Chem., 2015, 290, 8949–8963 CrossRef CAS PubMed.
- D. Xu, R. E. Perez, M. H. Rezaiekhaligh, M. Bourdi and W. E. Truog, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2009, 297, L44–L51 CrossRef CAS PubMed.
- M. Corazzari, P. E. Lovat, J. L. Armstrong, G. M. Fimia, D. S. Hill, M. Birch-Machin, C. P. Redfern and M. Piacentini, Br. J. Cancer, 2007, 96, 1062–1071 CrossRef CAS PubMed.
- I. Nemere, N. Garbi and Q. Winger, J. Cell. Biochem., 2015, 116, 380–385 CrossRef CAS PubMed.
- L. Wang, Y. Wu, J. Zhou, S. S. Ahmad, B. Mutus, N. Garbi, G. Hammerling, J. Liu and D. W. Essex, Blood, 2013, 122, 3642–3650 CrossRef CAS PubMed.
- Y. Wu, S. S. Ahmad, J. Zhou, L. Wang, M. P. Cully and D. W. Essex, Blood, 2012, 119, 1737–1746 CrossRef CAS PubMed.
- Q. Cui, Y. Chen, M. Zhang, L. Shan, Y. Sun, P. Yu, G. Zhang, D. Wang, Z. Zhao, Q. Xu, B. Xu and Y. Wang, Chem. Biol. Drug Des., 2014, 84, 282–291 CAS.
- G. Cui, L. Shan, M. Hung, S. Lei, I. Choi, Z. Zhang, P. Yu, P. Hoi, Y. Wang and S. M. Lee, Int. J. Cardiol., 2013, 168, 1349–1359 CrossRef PubMed.
- I. V. Shilov, S. L. Seymour, A. A. Patel, A. Loboda, W. H. Tang, S. P. Keating, C. L. Hunter, L. M. Nuwaysir and D. A. Schaeffer, Mol. Cell. Proteomics, 2007, 6, 1638–1655 CAS.
- R. Jasuja, F. H. Passam, D. R. Kennedy, S. H. Kim, L. van Hessem, L. Lin, S. R. Bowley, S. S. Joshi, J. R. Dilks, B. Furie, B. C. Furie and R. Flaumenhaft, J. Clin. Invest., 2012, 122, 2104–2113 CAS.
- N. Dickerhof, T. Kleffmann, R. Jack and S. McCormick, FEBS J., 2011, 278, 2034–2043 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 Search PubMed.
- K. Fukuda, S. S. Davies, T. Nakajima, B. H. Ong, S. Kupershmidt, J. Fessel, V. Amarnath, M. E. Anderson, P. A. Boyden, P. C. Viswanathan, L. J. Roberts and J. R. Balser, Circ. Res., 2005, 97, 1262–1269 CrossRef CAS PubMed.
- F. R. Laurindo, L. A. Pescatore and C. Fernandes Dde, Free Radical Biol. Med., 2012, 52, 1954–1969 CrossRef CAS PubMed.
- L. A. Loeb and R. W. Gross, J. Biol. Chem., 1986, 261, 467–470 Search PubMed.
- S. Wagner, A. G. Rokita, M. E. Anderson and L. S. Maier, Antioxid. Redox Signaling, 2013, 18, 1063–1077 CrossRef CAS PubMed.
- U. A. Bommer and B. J. Thiele, Int. J. Biochem. Cell Biol., 2004, 36, 379–385 CrossRef CAS.
- D. van der Vlies, E. H. Pap, J. A. Post, J. E. Celis and K. W. Wirtz, Biochem. J., 2002, 366, 825–830 CrossRef CAS PubMed.
- G. Cui, L. Shan, L. Guo, I. K. Chu, G. Li, Q. Quan, Y. Zhao, C. M. Chong, Z. Zhang, P. Yu, M. P. Hoi, Y. Sun, Y. Wang and S. M. Lee, Sci. Rep., 2015, 5, 10353, DOI:10.1038/srep10353.
- L. Ellgaard and L. W. Ruddock, EMBO Rep., 2005, 6, 28–32 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra08551a |
| ‡ These two authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.