
Enhancement of the sensitivity of valacyclovir and acyclovir for their spectrofluorimetric determination in human plasma†
Abstract
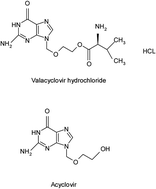
Two rapid, simple and highly sensitive spectrofluorimetric methods have been developed and validated for determination of valacyclovir hydrochloride (VAC) and acyclovir (ACV). The first method is based on measuring the intrinsic fluorescence of VAC or ACV in an aqueous acidic medium (pH 1.3) at 370 nm after excitation at 280 nm. The fluorescence intensity–concentration plots of VAC and ACV were rectilinear over the concentration ranges of 0.4–5.0 and 0.3–4.0 μg ml−1, respectively. The second method was based on the enhancement of the fluorescence intensity using sodium dodecyl sulfate (SDS) as a micellar system in an aqueous acidic medium (pH 1.3). This is the first attempt for enhancement of the sensitivity of these drugs by spectrofluorimetry. The addition of 2% w/v SDS, produced about 2.7 and 2.9 fold enhancements in the relative fluorescence intensity of VAC and ACV, respectively. The linear range for the second method was 0.2–2.5 and 0.1–1.25 μg ml−1, respectively. The proposed methods were successfully applied for determination of VAC and ACV in pharmaceutical preparations without interference from the common excipients. The high sensitivity of the micellar method permits its application for determination of ACV in human plasma with good percentage recovery.
 Please wait while we load your content...
Please wait while we load your content...

