
Lipase catalyzed desymmetrization of roof shape cis-11,12-bis(hydroxymethyl)-9,10-dihydro-9,10-ethanoanthracene†
Nilesh Jain and
Ashutosh V. Bedekar*
Department of Chemistry, Faculty of Science, M.S. University of Baroda, Vadodara 390 002, India. E-mail: avbedekar@yahoo.co.in; Tel: +91-265-2795552
First published on 15th July 2015
Abstract
Biocatalyzed desymmetrization of roof shape meso cis-11,12-bis(hydroxymethyl)-9,10-dihydro-9,10-ethanoanthracene has been achieved. The absolute configuration of the product is established by single crystal X-ray analysis of its diastereomer prepared with R-chloropropionic acid. Single crystal analysis of the chiral roof shape monoacetate shows a P-helical motif due to the extended intramolecular hydrogen bonding while the racemic sample exhibits intermolecular double hydrogen bonding holding two molecules together as a dimmer.
Introduction
The synthesis of chiral molecules with different shapes and sizes in optically pure form is an important aspect of modern organic chemistry. Applications of chiral molecules, natural and unnatural, are found in wide range of areas covering medicinal, supramolecular, materials, analytical chemistry, agro and food industry and in the asymmetric synthesis of other chiral compounds. The strategy of chiral pool synthesis is widely explored to access chiral starting materials for useful synthesis,1 but it has its limitations. Scope of applications of unnatural chiral compounds is widely being recognized. Recent focus is on the use of efficient synthetic methodologies to access such chiral molecules from achiral or prochiral materials. Along with several elegant methods of generation of chiral elements by synthetic operations, it is also possible to separate enantiomers of the chiral compounds. Separation of isomers of chiral compounds can be achieved by metal based catalytic reactions or by non-metal based organocatalytic and enzymatic reactions. Enzymes are generally recognized as effective, mild, selective, environment friendly catalysts for accessing optically active compounds, mostly by separating the isomers.2 The development is also helped by availability of immobilized enzymes which can tolerate non-aqueous reaction conditions for practical applications.3 Although biocatalysts can target several functional groups like esters, acids, amines they are often studied for effecting conversion of alcohols to esters by suitable transesterification reactions. Racemic alcohols can be selectively converted to the esters of one enantiomer while keeping the other unaffected, based on the kinetic resolution process.4 The drawback of limited yield, maximum 50%, of the product and unchanged starting material in this process is overcome by dynamic kinetic resolution5 which consists of carrying out continuous interconversion or racemization of the substrate molecule. At the same time the process of desymmetrization which effectively removes the element of symmetry from suitable molecules is an attractive option to access chiral molecules. Basically molecules with two identical functional groups are subjected to selective conversion of one of them to different functionality, theoretically giving quantitative yield of the product. Such desymmetrization has been known to be achieved by metal catalytic or non-enzymatic reactions6 or by biocatalyzed conversions.7 The protocol of biocatalyzed desymmetrization has been applied on meso-diols for the preparation of chiral molecules8 and for the synthesis of complex natural products.9 The most common arrangement of meso-diol A and B which can be subjected to enzymatic desymmetrization are presented in Fig. 1. | ||
| Fig. 1 Basic types of meso diols. | ||
Some molecules which posses shape similar to a roof of a hut or a house, called as “Roof Shape” molecules, were introduced by Weber for the study of inclusion properties as clathrate hosts.10 Similarly other known molecules which are structurally analogues, such as iptacene,11 triptacene,12 and molecular tweezers13 have shown useful applications. Structurally similar roof shape molecules have been well studied for their various properties and applications.14 The roof shape molecules are synthesized by Diels–Alder cycloaddition of anthracene with electron deficient dienophile. Further functional group modifications have lead to a number such derivatives with wide applications in fields such as medicinal chemistry,15 as ligands in catalytic transformations,16 for organocatalytic reactions,17 for preparation of chiral selectors,18 and in the synthesis of functional polymers.19 The optically pure isomers of such anthracene derived roof shape molecules have been obtained by asymmetric synthesis utilizing chiral fumaric acid derivatives16a,20 or by separating the enantiomers by fractional crystallization with chiral resolving agents.21
In our continuing interests in the roof shape molecules we have prepared and resolved alcohol 1 and diol 2 by enzymatic selective esterification reactions (Chart 1).22 These molecules were prepared from anthracene by Diels–Alder cycloaddition reaction with suitable dienophiles such as methyl acrylate and dimethyl fumarate, followed by the reduction of the roof shape adducts. The alcohol 1 and diol 2 were subjected to enzymatic transesterification reaction with appropriate acylating agents in presence of biocatalysts. The optically pure alcohol and diol were then converted to a series of amines by simple chemical operations. The resulting optically pure roof shape amines were screened as chiral solvating agents22b for discrimination of signals in nuclear magnetic resonance spectroscopy.
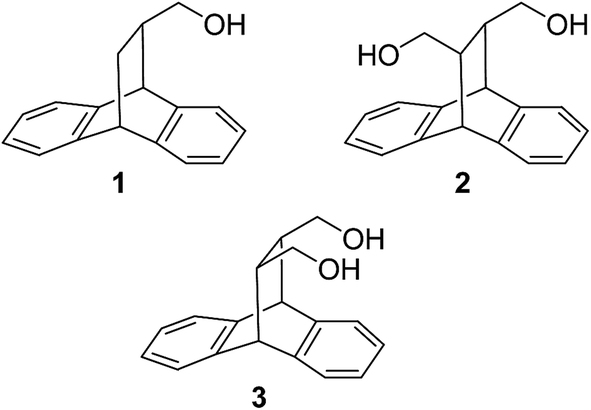 | ||
| Chart 1 Roof shape alcohol and diols. | ||
In the present study we extend the scope of such molecules by studying the meso isomer of cis-diol 3, which was synthesized and resolved using biocatalytic approach. The separation of cis-diol 3 involving enantioselective acylation and hence may give theoretically up to quantitative yield as against the case of trans-diol 2.
Result and discussion
The synthesis of cis-diol 3 is outlined in Scheme 1. The roof shape framework is constructed by Lewis acid catalyzed Diels–Alder cycloaddition of anthracene and maleic anhydride at ambient conditions. The adduct anhydride 4 separated by crystallization was then subjected to reduction under mild conditions23 of I2–NaBH4 to afford cis-diol 3 in good yield.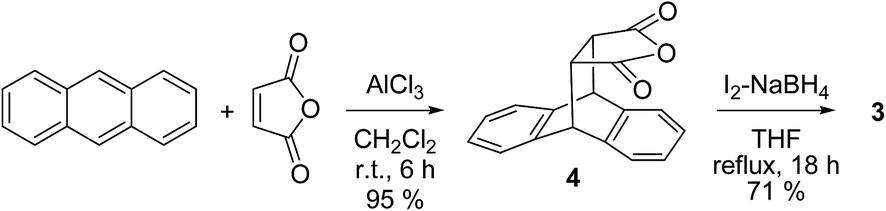 | ||
| Scheme 1 Synthesis of meso diol 3. | ||
The cis-diol 3 was then subjected to desymmetrization reaction involving enzyme mediated transesterification reaction (Scheme 2). In our earlier study on resolution of roof shape alcohols we had successfully screened Novozym-435 for alcohol 1,22b while steapsin lipase was effective for trans-diol 222a and THF was found to be a good solvent for both reactions. The optically pure materials were obtained in high purity and efficiency, particularly at low temperature. Our observation of higher selectivity at lower temperature is consistent with the literature reports.24 Hence, we started our initial screening with these conditions for the desymmetrization of diol 3 (Table 1).
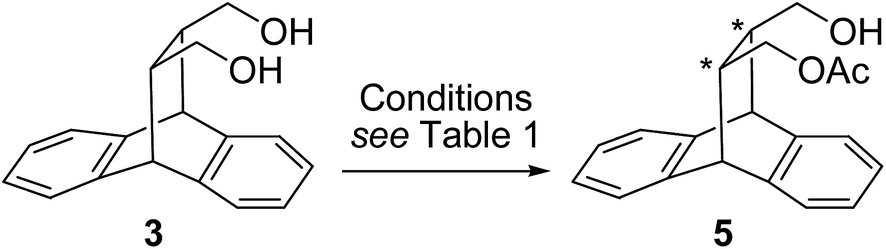 | ||
| Scheme 2 Desymmetrization of 3. | ||
| No. | Enzymeb | Solvent | Time (h) | Acyl donorc (eq.) | Yieldd (%) [eee (%)] of 5 |
|---|---|---|---|---|---|
| a All reaction run at 8–9 °C.b SL = steapsin lipase; N-435 = Novozym-435; CR = Candida rugosa.c VA = vinyl acetate; IPA = isopropenyl acetate; EA = ethyl acetate; BA = butyl acetate.d Isolated.e Determined by HPLC on Chiralpak-ODH.f As solvent.g No reaction. | |||||
| 1 | SL | THF | 72 | VA (3) | 35 [13] |
| 2 | SL | THF | 72 | VA (15) | 71 [48] |
| 3 | SL | THF | 60 | VA (30) | 93 [72] |
| 4 | SL | THF | 30 | VA (60) | 90 [79] |
| 5 | N-435 | THF | 60 | VA (7) | 29 [0] |
| 6 | N-435 | THF | 60 | VA (7) | 35 [0] |
| 7 | CR | THF | 60 | VA (7) | 12 [0] |
| 8 | SL | DME | 60 | VA (30) | 75 [55] |
| 9 | SL | Toluene | 60 | VA (30) | 53 [55] |
| 10 | SL | THF | 120 | EAf | 27 [71] |
| 11 | SL | THF | 60 | IPA (30) | 35 [52] |
| 12 | SL | THF | 60 | BA (30) | — [—]g |
The reaction of diol 3 with vinyl acetate as acyl donor in presence of steapsin lipase in THF was clean and only one product of mono acylation 5 was detected in the reaction mixture. The product could be easily separated and analyzed. The optical purity of the product 5 was established by HPLC analysis on Chiralpak-ODH column. As can be observed acylation was more effective with higher amount of acylating reagent. Comparison of amount of vinyl acetate and the reaction time indicated the optimum condition was achieved with its use in large excess and much shorter reaction time (entry 4). The other enzymes Novozym-435 and Candida rugosa were ineffective in chiral discrimination. We scanned two more solvents, DME and toluene, although conversion was notable the selectivity was moderate (entry 8 & 9). We also scanned different acyl donors, where ethyl acetate was found effective when used as solvent. Although the selectivity was good the conversion was quite poor (entry 10 to 12).
The enzyme mediated reactions on structurally demanding substrates are often found difficult and challenging.8b,8c,25 The present cis-diol 3 in comparison with the other two roof shape substrates 1 and 2 may also pose difficulty due to the similar considerations. This was addressed by studying the increased ratio of enzyme to substrate (Table 2). Although there was not much improvement in the conversion, the selectivity was considerably improved when high amounts were used with excess of acyl donor. Significantly no product of diacetylation was observed in any of the experiments investigated in the present study.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :enzyme (steapsin lipase) ratio
:enzyme (steapsin lipase) ratio
Molecules pose some additional issues where the two groups capable of chemically undergoing acylation reactions are juxtaposed in the manner where the internal acyl transfer reactions are feasible. When the two alcohol groups are placed close to each other the acyl group may migrate leading to the interconversion of the mono acyl derivative. Such problem leads to low selectivity as discussed by Fadel and Arzel8b and others.8c,26 In the course of our present study we envisaged similar possibility as the two hydroxymethyl arms in cis-diol 3 are protruding in the same direction and are placed close to each other. The possibility may attribute to the observed low selectivity in some of the experiments we had studied. To confirm this possibility a sample of optically pure mono acetate 5 was refluxed in isopropanol and its purity was checked periodically (Table 3). Within a relatively short time the product lost its optical purity to a considerable extent confirming the isomerization of mono acetate 5. The possible mode of internal transfer of acyl unit in 5 may be explained in Scheme 3. Attack of free hydroxyl group on the acetate can give a meso intermediate 1,3-dioxolan-2-ol derivative 6, which can open from two sides, ‘path-a’ giving one isomer 5a, while ‘path-b’ may lead to the other isomer 5b, both the steps can be promoted at higher temperature.
 | ||
| Scheme 3 Mode of racemization of 5. | ||
Optically active compounds, particularly with more than one chiral center, are often subjected to enrichment by recrystallization under proper conditions. Right from the pioneering work of Louis Pasteur such observations are well studied.27 The procedure of enrichment of chiral isomers during the process of crystallization appears to be more of an art than science and success depends on certain structural arrangement responsible for definite supramolecular interactions. There are frequent reports in the literature where the technique of recrystallization has been utilized to improve the optical purity of the compounds.28 The isolated optically enriched sample of the product 5 was crystallized from different solvents in order to purify as well as to further improve the ratio of its diastereomers. Different solvents were investigated to recrystallize optically pure samples of 5 (∼70% ee). In dichoromethane–hexane mixture (10:90) we observed formation of crystals of two distinct shapes. Careful observation indicated that few crystals were of plate like shape and more transparent in nature. These crystals, though seen in very few numbers, were physically separated and characterized by single crystal X-ray diffraction analysis and their optical purity was established by chiral phase HPLC analysis (Chiralpak ODH). The optical purity was observed to be enriched and on average seen to be in the rage of 93–95%. The X-ray analysis of these crystals showed unique features (Fig. 2).29 The crystal showed packing of only one stereoisomer with a chiral space group of P212121. Moreover intermolecular hydrogen bonding between the carbonyl oxygen of –OC(O)CH3 of one molecule and the hydroxyl group of the other was observed. The hydrogen bond distance was found to be 2.138 Å, typical of such a case.
 | ||
| Fig. 2 ORTEP Diagram of optically pure mono acetate 5. | ||
The intermolecular hydrogen bonding in the optically pure crystal of 5 was observed in an extended linear way between the series of molecules (Fig. 3). Moreover it is noteworthy to see a P-helical motif along with the a axis (marked by yellow rod in Fig. 3) with a helical pitch of 15.782 Å. Such molecular assembly formed due to weak hydrogen bond network to form a helix with particular stereoisomer is known in the literature.30
 | ||
| Fig. 3 Diagram showing topology formed in crystal due to extended intermolecular H-bond network for optically pure mono acetate 5. Right side is space filled picture where the alternate molecules are shown in green and blue colour. | ||
However, the majority of crystals of 5 were colorless and more like cube shape. These crystals were racemic in nature, established by specific optical rotation as well as by HPLC analysis. The single crystal analysis indicated a completely different arrangement (Fig. 4).29 The analysis revealed the unit cell was consisting of two molecules of opposite stereochemistry held together by two sets of intermolecular hydrogen bonding between the same functionalities. The hydrogen bond distance was seen to be 2.308 Å in the case of racemic sample. Slightly weaker hydrogen bond in racemic sample may be attributed to quite a large macrocycle of eighteen atoms. The space group of the racemic crystal was observed to be P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and a well organized dimmeric arrangement was detected. No extended hydrogen bond network was observed in the crystal of racemic mono acetate 5. Such contrasting crystal behavior between the optically pure and racemic sample of same molecule is a rare phenomena. However, the recrystallization of initial racemic sample of 5 in various solvents under different conditions yielded racemic crystals; hence the possibility of spontaneous resolution31 may be low. It was also observed that the melting points of the chiral and racemic samples of 5 differ, as also observed earlier for similar roof shape trans-diol.20a The thermal analysis TG-DTA indicated the m.p. of chiral 5 (sample of 95% ee) was 104.5 °C while the racemic sample melted at higher temperature of 119.3 °C.
and a well organized dimmeric arrangement was detected. No extended hydrogen bond network was observed in the crystal of racemic mono acetate 5. Such contrasting crystal behavior between the optically pure and racemic sample of same molecule is a rare phenomena. However, the recrystallization of initial racemic sample of 5 in various solvents under different conditions yielded racemic crystals; hence the possibility of spontaneous resolution31 may be low. It was also observed that the melting points of the chiral and racemic samples of 5 differ, as also observed earlier for similar roof shape trans-diol.20a The thermal analysis TG-DTA indicated the m.p. of chiral 5 (sample of 95% ee) was 104.5 °C while the racemic sample melted at higher temperature of 119.3 °C.
 | ||
| Fig. 4 ORTEP Diagram of racemic mono acetate 5 showing dimmer formation. | ||
The absolute configuration of enantiomerically enriched sample of 5 was established by preparing its ester with (R)-2-chloropropionic acid. The chirally pure mono acetate 5 obtained during the resolution was converted to the diastereomerically pure ester 7 (Scheme 4). The single crystal X-ray diffraction analysis29 of 7 clearly established the absolute configuration of the stereogenic carbons of the original sample of 5 (Fig. 5).
 | ||
| Scheme 4 Conversion of 5 to ester of known absolute configuration. | ||
 | ||
| Fig. 5 ORTEP Diagram of 7. | ||
The present roof shape chiral molecules are good candidates to be used in asymmetric synthesis, catalysis and in molecular recognition studies. To increase their utility we need to develop their conversion to other functional groups. Our studies have also established the cis-isomers undergoes loss of its optical purity due to the internal acyl transfer process. In order to address these issues we consider conversion the free hydroxyl group of optically pure sample of mono acetate 5a to its azide by Mitsunobu reaction with DPPA.32 We followed the standard procedure33 to convert sample of 5a with optical purity of 79% ee under ultrasonic irradiation to afford azido-acetate 8, which was converted to azido-alcohol 9 by acid catalyzed hydrolysis. The optical purity of 9 remained unchanged during the procedure as both the products were stable and no isomerisation was observed (Scheme 5).
 | ||
| Scheme 5 Synthesis of azido-acetate 8 and azido-alcohol 9. | ||
In the earlier part of the study we have established the thermal isomerisation of optically pure sample of mono-acetate 5a (Table 3). During the course of work-up and purification by column chromatography, concentration of the fractions etc. there was a chance of isomerisation of the compound and may contribute in the lowering of optical purity. In order to ascertain this aspect we performed a separate set of lipase mediated resolution of cis-diol 3, where the crude sample, after just filtration of the immobilized enzyme, was subjected to the above sequence of Mitsunobu reaction and hydrolysis (Scheme 6). The final product 9 was purified by column chromatography and analyzed for its optical purity. The product 9 obtained here showed higher optical purity (85% ee) and the process can be extended for practical applications.
 | ||
| Scheme 6 One step synthesis of azido-alcohol 9. | ||
The conversion of meso cis-diol 3 to optically pure azido-alcohol 9 will open further possibilities to study applications of functionalized roof shape chiral compounds.
Experimental section
Thin layer chromatography was performed on silica gel plates quoted on aluminium sheets. The spots were visualized under UV light or with iodine vapor. All the compounds were purified by column chromatography on silica gel (60–120 mesh). All reactions were carried out under an inert atmosphere (nitrogen) unless other conditions are specified. NMR Spectra were recorded on Bruker Avance 400 Spectrometer (400 MHz for 1H NMR, 100 MHz for 13C NMR) with CDCl3 as solvent and TMS as internal standard. Mass spectra were recorded on Thermo-Fischer DSQ II GCMS instrument; IR spectra were recorded on a Perkin-Elmer FTIR RXI spectrometer as KBr pallets. Melting points were recorded in Thiele's tube using paraffin oil and are uncorrected.9,10-Dihydroanthracene-9,10-α,β-succinic acid anhydride (4)
To a suspension of anthracene (10.0 g, 56.2 mmol) in dry dichloromethane (100 mL) under nitrogen atmosphere kept in ice bath (0 to 3 °C), aluminum chloride (7.47 g, 56.2 mmol) was added portion wise. After stirring for 15 min, maleic anhydride (5.5 g, 56.2 mmol) was added. The resulting mixture was stirred at room temperature (6 h), and poured into ice-water (200 mL), the separated organic layer was collected and the aqueous layer was extracted with dichloromethane (3 × 150 mL). The combined organic extracts were washed with water (2 × 50 mL), dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by crystallization with ethyl acetate (14.2 g, 91%).M.p. = 255 °C (lit.34 = 257–258 °C). 1H NMR (400 MHz, CDCl3): δ 3.557 (s, 2H) 4.854 (s, 2H), 7.21–7.24 (m, 4H), 7.34–7.42 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 47.4, 47.9, 124.4, 125.2, 127.2, 127.7, 138.1, 140.6, 170.5. IR (KBr): ν 2977, 1783, 1706, 1463, 1258, 1065, 751 cm−1.
cis-11,12-Bis(hydroxymethyl)-9,10-dihydro-9,10-ethanoanthracene (3)
An assembly of three necked round bottom 500 mL flask with magnetic stir bar, a reflux condenser and an addition funnel is prepared. The flask was charged with sodium borohydride (2.7 g, 72.4 mmol) and dry THF (50 mL). To this mixture kept at 0 °C, the above anhydride (4) (5.0 g, 18.1 mmol) was added in one portion. A solution of iodine (9.2 g, 36.2 mmol dissolved in 50 mL THF), was added drop wise over 30 min resulting in vigorous evolution of hydrogen. After addition of iodine was complete the flask was heated to reflux (18 h) and then cooled to room temperature. Dry methanol was added cautiously, until the mixture became clear. After stirring for 30 min, the solvent was removed by rotary evaporation leaving a white paste which was dissolved by addition of aqueous KOH solution (5%, 150 mL) was stirred (1 h) and extracted with ethyl acetate (3 × 250 mL). The organic extracts were dried over anhydrous sodium sulfate and concentrated in vacuum, which was further purified by column chromatography on silica gel (CH2Cl2/MeOH, gradient elution: 95/5). Rf = 0.4 (CH2Cl2–MeOH, 4:1) (3.81 g, 71%).
M.p. 224 °C (lit.35 = 222–225 °C). 1H NMR (400 MHz, CDCl3): 2.35–2.42 (m, 2H), 2.67 (br signal, 2H), 3.28–3.36 (dd, 11.2, 4.0 Hz, 2H), 3.55–3.58 (dd, J = 12.2, 4.0 Hz, 2H), 4.21 (s, 2H), 7.01–7.13 (m, 4H), 7.20–7.31 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 43.5, 47.9, 64.4, 123.3, 124.7, 125.8, 125.9, 140.8, 143.4. IR (KBr): ν 3325, 3235, 3017, 1466, 1359, 1040, 1022, 749 cm−1.
:1).cis-9,10-Dihydro-9,10-ethanoanthracene-11-acetoxymethyl-12-methanol (5)
M.p. = 119.3 °C for racemic, 104.5 °C for (11R, 12S). [α]28D = 30.4 (c = 0.5, MeOH for 95% ee). HPLC condition Chiralpak OD-H column: 10% IPA-Hexane, UV = 254 nm, flow = 0.8 mL min−1, Rt = 15.8 min (1st peak) [11R, 12S-isomer] and Rt – 18.1 min (2nd peak) [11S, 12R isomer]. 1H NMR (400 MHz, CDCl3): δ 2.12 (s, 3H), 2.31–2.44 (m, 2H), 3.13–3.17 (dd, J = 9.2, 5.6 Hz, 1H), 3.49–3.60 (m, 2H), 3.93–3.97 (dd, J = 10.8, 5.6 Hz), 4.32 (d, J = 2 Hz, 1H), 4.45 (d, J = 1.8 Hz, 1H), 7.13–7.27 (m, 4H), 7.28–7.33 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 21.1, 39.6, 43.1, 46.2, 46.7, 62.3, 64.3, 123.5 (2C), 125.2, 125.3, 125.8, 126.0, 126.1 (2C), 140.5, 140.8, 143.1, 143.5, 170.9. IR (KBr): ν 3356, 3021, 2957, 1724, 1468, 1368, 1233, 1020, 746 cm−1. HRMS: HRMS (ESI+) m/z calculated for C20H20O3 [M + Na]+ 331.1304; found 331.1305.cis-9,10-Dihydro-(9,10-ethanoanthracene-11-yl)methyl 2-chloropropanoate acetoxymethyl-12-acetoxymethyl (7)
Alcohol-(5) (0.1 g, 0.32 mmol), DCC (0.066 g, 0.32 mmol) was placed in two necked flask under N2 atmosphere, dissolved in 5 mL of dichloromethane and cooled (0 °C). A solution of (R)-2-chloro propionic acid (0.027 mL, 0.32 mmol) in dichloromethane (2 mL) was added drop wise. The reaction mixture was stirred (0 °C, 3 h). After the reaction was over the reaction mixture was passed through celite, washed with dichloromethane and purified by column chromatography on silica gel (PE/EtOAc, gradient elution: 90/10). Rf = 0.7 (PE/EtOAc, 3:1). White solid (0.09 g, 71%).
M.p. = 126 °C, [α]28D = 19.7 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ 1.74 (d, J = 6.8 Hz, 3H), 2.10 (s, 3H), 2.42–2.53 (m, 2H), 3.61–3.75 (m, 2H), 3.88–3.97 (dd, J = 11.2, 5.6 Hz, 1H), 4.02–4.05 (dd, J = 10.8, 5.4 Hz, 1H), 4.32 (d, J = 2 Hz, 1H), 4.37 (d, J = 2 Hz, 1H), 4.43–4.49 (q, J = 6.8 Hz, 1H), 7.14–7.17 (m, 4H), 7.26–7.33 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 21.1, 21.3, 39.6, 39.7, 46.3, 46.6, 52.4, 64.0, 65.5, 123.6, 123.7, 125.4, 125.8, 126.1 (2C), 126.3 (2C), 140.1, 140.2, 142.8, 143.0, 169.7, 170.7. IR (KBr): ν 2964, 1740, 1696, 1468, 1274, 1234, 1061, 757, 550 cm−1. HRMS (ESI+) m/z calculated for C23H23ClO4 [M + Na]+ 421.1183; found 421.1177.
cis-9,10-Dihydro-(9,10-ethanoanthracene-11-(azidomethyl)-12-yl)methy acetate (8)
A solution of PPh3 (0.236 g, 0.90 mmol) in dry THF (1.5 mL) was cooled (0 °C) under N2 atmosphere. To the mixture deithylazodicarboxlate (0.14 mL, 0.90 mmol) was slowly added. Then solution of monoacetate (5) (0.16 g, 0.60 mmol) in dry THF (2 mL) was added to the reaction mixture, which was stirred (10 min. at 0 °C). To this DPPA (0.16 mL, 0.75 mmol) was added drop wise. The reaction mixture was then exposed to ultrasonic irradiation (30 min). The solvent was removed under reduced pressure and the crude material was purified silica gel column chromatography on silica gel (PE/EtOAc, gradient elution: 95/5). Rf = 0.78 (Pet Ether/EtOAc, 4:1).
White solid (0.103 g, 60%). M.p. = 110–111 °C, [α]28D = 11.01 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ 2.11 (s, 3H), 2.32–2.42 (m, 2H), 2.68–2.72 (dd, J = 10.8, 4.8 Hz, 1H) 3.29–3.34 (dd, J = 9.6, 4.8 Hz, 1H), 3.60–3.65 (dd, 9.6, 8.4 Hz, 1H), 3.78–3.83 (dd, J = 10.8, 6.0 Hz, 1H), 4.29 (d, J = 2 Hz, 1H), 4.41 (d, J = 2 Hz, 1H), 7.14–7.28 (m, 2H), 7.30–7.35 (m, 4H). 13C NMR (100 MHz, CDCl3): 21.0, 39.8, 40.3, 46.5, 46.7, 51.5, 64.1, 123.5, 123.8, 125.4, 125.6, 126.1, 126.2, 126.3 (2C), 140.1, 140.3, 142.7, 142.9, 170.7. IR (KBr): ν 3024, 2099, 1734, 1468, 1235, 1035, 755 cm−1. HRMS (ESI+) m/z calculated for found C20H19N3O2 [M + Na]+ 356.1369; found 356.1370.
cis-9,10-Dihydro-(9,10-ethanoanthracene-11-(azidomethyl)-12-yl)methanol (9)
To a solution of 8 (0.1 g, 0.3 mmol) in methanol (5 mL), HCl (0.04 mL, 0.5 mmol, 36%) was added. The reaction mixture was refluxed (2 h). After completion of reaction (TLC), MeOH was evaporated under reduce pressure. The residue was taken in ethyl acetate (25 mL) and washed with water (2 × 15 mL). The organic layer was dried with sodium sulfate and concentrated to afford (9) as white solid 0.079 g (90%). Rf = 0.60 (Pet Ether/EtOAc, 4:1).
M.p. = 135 °C, [α]28D = 29.0 (c = 0.5, CHCl3 for 85% ee). HPLC Condition Chiralpak OD-H column: 20% IPA-hexane, UV = 215 nm, flow = 0.75 mL min−1, Rt – 8.7 min (1st peak) and Rt – 15.7 min (2nd peak). 1H NMR (400 MHz, CDCl3): δ 2.31–2.35 (m, 2H), 2.75–2.80 (m, 1H), 3.20–3.24 (m, 1H), 3.33–3.41 (m, 2H), 4.39 (s, 2H) 7.14–7.34 (m, 4H), 7.29–7.34 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 40.4, 43.3, 46.6, 46.9, 51.2, 62.7, 123.5, 123.7, 125.1, 125.5, 125.9, 126.1, 140.3, 140.8, 142.8, 143.4. IR (KBr): ν 3370, 3041, 2933, 2099, 1468, 1273, 1002, 750 cm−1. HRMS (ESI+) m/z calculated for C18H17N3O [M + Na]+ 314.1269; found 314.1264.
Conclusions
Thus we have presented our efforts to resolve the meso isomer of novel roof shape cis-11,12-bis(hydroxymethyl)-9,10-dihydro-9,10-ethanoanthracene by enzyme mediated desymmetrization. The absolute configuration of the product was established by single crystal X-ray analysis of its diastereomer prepared with R-chloropropionic acid. Single crystal analysis of the chiral roof shape monoacetate shows an interesting helical motif of P-conformation due to the intramolecular hydrogen bonding. Similar arrangement was not seen in the racemic sample of the same compound, where its two molecules of opposite chirality were nicely held together by intermolecular hydrogen bonding.Acknowledgements
We thank Council of Scientific and Industrial Research (CSIR), New Delhi for the award of Senior Research Fellowship to NJ. We are also grateful to DST-PURSE for the single crystal X-ray diffraction facility of the Faculty of Science.References and Notes
- (a) H. U. Blaser, Chem. Rev., 1992, 92, 935–952 CrossRef CAS; (b) S. Haleema, P. V. Sasi, I. Ibnusaud, P. L. Polavarapu and H. B. Kagan, RSC Adv., 2012, 2, 9257–9285 RSC; (c) P. Singh, K. Samanta, S. K. Das and G. Panda, Org. Biomol. Chem., 2014, 12, 6297–6339 RSC; (d) C. Bhat and S. G. Tilve, RSC Adv., 2014, 4, 5405–5452 RSC.
- (a) R. D. Schmid and R. Verger, Angew. Chem., Int. Ed., 1998, 37, 1608 CrossRef; (b) S. M. Roberts, J. Chem. Soc., Perkin Trans. 1, 1999, 1–21 RSC; (c) R. P. Patel, Stereoselective biocatalysis, Marcel Dekker, New York, 2000 Search PubMed; (d) A. S. Bommarius and B. R. Riebel, Biocatalysis, Wiley-VCH, Weinheim, 2004 Search PubMed; (e) K. Faber, Biotransformations in organic chemistry, Springer-Verlag, Heidelberg, 2004 Search PubMed.
- (a) G. Carrea and S. Riva, Angew. Chem., Int. Ed., 2000, 39, 2226 CrossRef CAS; (b) A. M. Klibanov, Nature, 2001, 409, 241 CrossRef CAS PubMed; (c) L. Cao, L. van Langen and R. A. Sheldon, Curr. Opin. Biotechnol., 2003, 14, 387 CrossRef CAS; (d) G. Carrea and S. Riva, Organic synthesis with enzymes in non-aqueous media, Wiley-VCH, Weinheim, 2008 Search PubMed.
- (a) E. Schoffers, A. Golebiowski and C. R. Johnson, Tetrahedron, 1996, 52, 3769–3826 CrossRef CAS; (b) A. Ghanem and H. Y. Aboul-Enein, Chirality, 2005, 17, 1–15 CrossRef CAS PubMed.
- O. Pàmies and J.-E. Bäckvall, Chem. Rev., 2003, 103, 3247 CrossRef PubMed.
- (a) T. Harada, K. Sekiguchi, T. Nakamura, J. Suzuki and A. Oku, Org. Lett., 2001, 3, 3309–3312 CrossRef CAS PubMed; (b) A. J. Blake, S. C. Hume, W.-S. Li and N. S. Simpkins, Tetrahedron, 2002, 58, 4589–4602 CrossRef CAS; (c) S. E. de Sousa, P. O'Brien and C. D. Pilgram, Tetrahedron, 2002, 58, 4643–4654 CrossRef CAS; (d) Y. Matsumura, T. Maki, S. Murakami and O. Onumura, J. Am. Chem. Soc., 2003, 125, 2052–2053 CrossRef CAS PubMed; (e) H. Shimizu, S. Onitsuka, H. Egami and T. Katsuki, J. Am. Chem. Soc., 2005, 127, 5396–5413 CrossRef CAS PubMed; (f) V. B. Birman, H. Jiang and X. Li, Org. Lett., 2007, 9, 3237–3240 CrossRef CAS PubMed; (g) E. P. Kündig, A. Enriquez Garcia, T. Lomberget, P. Perez Garcia and P. Romanens, Chem. Commun., 2008, 3519–3521 RSC; (h) Y. Tsuda, M. Kuriyama and O. Onomura, Chem.–Eur. J., 2012, 18, 2481–2483 CrossRef CAS PubMed; (i) S. J. Canipa, A. Stute and P. O’Brien, Tetrahedron, 2014, 70, 7395–7403 CrossRef CAS PubMed.
- (a) E. García-Urdiales, I. Alfonso and V. Gotor, Chem. Rev., 2005, 105, 313–354 CrossRef PubMed; (b) For the update of this review, see E. García-Urdiales, I. Alfonso and V. Gotor, Chem. Rev., 2011, 111, PR110–PR180 CrossRef PubMed.
- Select examples of enzyme mediated desymmetrization of meso-diols: (a) G. Nocolosi, A. Patti, M. Piattelli and C. Sanfillippo, Tetrahedron: Asymmetry, 1994, 5, 283–288 CrossRef; (b) A. Fadel and P. Arzel, Tetrahedron: Asymmetry, 1997, 8, 283–291 CrossRef CAS; (c) S. Akai, T. Naka, T. Fujita, Y. Takebo and Y. Kita, Chem. Commun., 2000, 1461–1462 RSC; (d) I. Borreguero, J. M. Sánchez-Montero, J. V. Sinisterra, A. Rumbero, J. A. Hermoso and A. R. Alcántara, J. Mol. Catal. B: Enzym., 2001, 11, 1013–1024 CrossRef CAS; (e) C. Neri and J. M. J. Williams, Tetrahedron: Asymmetry, 2002, 13, 2197–2199 CrossRef CAS; (f) R. Chênavert, G. Courchesne and F. Jacques, Tetrahedron: Asymmetry, 2004, 15, 3587–3590 CrossRef PubMed; (g) D. Acetti, E. Brenna, C. Fuganti, F. G. Gatti and S. Serra, Tetrahedron Lett., 2009, 50, 2249–2251 CrossRef CAS PubMed; (h) C. M. Sapu, J.-E. Bäckvall and J. Deska, Angew. Chem., Int. Ed., 2011, 50, 9731–9734 CrossRef CAS PubMed; (i) G. Obame, H. Pellissier, N. Vanthuyne, J.-B. Bongui and G. Audran, Tetrahedron Lett., 2011, 52, 1082–1085 CrossRef CAS PubMed; (j) N. Ríos-Lombardía, V. Gotor-Fernández and V. Gotor, J. Org. Chem., 2011, 76, 811–819 CrossRef PubMed.
- For some important examples of complex products: (a) A. D. Lebsack, J. T. Link, L. E. Overman and B. A. Stearns, J. Am. Chem. Soc., 2002, 124, 9008–9009 CrossRef CAS PubMed; (b) M. Lautens, J. T. Colucci, S. Hiebert, N. D. Smith and G. Bouchain, Org. Lett., 2002, 4, 1879–1882 CrossRef CAS PubMed; (c) U. Zutter, H. Iding, P. Spurr and B. Wirz, J. Org. Chem., 2008, 73, 4895–4902 CrossRef CAS PubMed; (d) K. C. Nicolaou, H. Zhang, A. Ortiz and P. Dagneau, Angew. Chem., Int. Ed., 2008, 47, 8605–8610 CrossRef CAS PubMed; (e) M. Candy, G. Audran, H. Bienayme, C. Bressy and J.-M. Pons, Org. Lett., 2009, 11, 4950–4953 CrossRef CAS PubMed.
- (a) E. Weber, I. Csöregh, J. Ahrendt, S. Finge and M. Czugler, J. Org. Chem., 1988, 53, 5831–5839 CrossRef CAS; (b) E. Weber, T. Hens, O. Gallardo and I. Csöregh, J. Chem. Soc., Perkin Trans. 2, 1996, 737–745 RSC.
- (a) J. Bouffard, R. F. Eaton, P. Muller and T. M. Swager, J. Org. Chem., 2007, 72, 10166–10180 CrossRef CAS PubMed; (b) T. M. Swager, Acc. Chem. Res., 2008, 41, 1181–1189 CrossRef CAS PubMed.
- L. Zhao, Z. Li and T. Worth, Chem. Lett., 2010, 39, 658–667 CrossRef CAS.
- M. Marquis, K. Kulikiewicz, S. Lebedkin, M. M. Kappes, C. Mioskowski, S. Meunier and A. Wagner, Chem.–Eur. J., 2009, 15, 11187–11196 CrossRef PubMed.
- (a) J. Karolak-Wojciechowska, H. B. Trzeźwińska, S. Alibert-Franco, C. Santelli-Rouvier and J. Barbe, J. Chem. Crystallogr., 1998, 28, 905–911 CrossRef CAS; (b) M. Smet, D. Corens, L. van Meervelt and W. Dehaen, Molecules, 2000, 5, 179–188 CrossRef CAS PubMed; (c) J. C. C. Atherton and S. Jones, Tetrahedron, 2003, 59, 9039–9057 CrossRef CAS PubMed.
- (a) S. Alibert, C. Santelli-Rouvier, B. Pradines, C. Houdoin, D. Parzy, J. Karolak-Wojciechowska and J. Barbe, J. Med. Chem., 2002, 45, 3195–3209 CrossRef CAS PubMed; (b) S. Alibert, C. Santelli-Rouvier, M. Castaing, M. Berthelot, G. Spengler, J. Molnar and J. Barbe, Eur. J. Med. Chem., 2003, 38, 253–263 CrossRef CAS; (c) J. Millet, M. Torrentino-Madamet, S. Alibert, C. Rogier, C. Santelli-Rouvier, J. Mosnier, E. Baret, J. Barbe, D. Parzy and B. Pradines, Antimicrob. Agents Chemother., 2004, 48, 2753–2756 CrossRef CAS PubMed.
- (a) H. Waldmann, M. Weigerding, C. Dreisbach and C. Wandrey, Helv. Chim. Acta, 1994, 77, 2111–2116 CrossRef CAS PubMed; (b) J. R. Scheffer and H. Ihmels, Liebigs Ann./Recl., 1997, 1925–1929 CrossRef CAS PubMed; (c) S. Hoshimoto, H. Matsunaga and T. Kunieda, Chem. Pharm. Bull., 2000, 48, 1541–1544 CrossRef CAS; (d) S. Doherty, E. G. Robins, J. G. Knight, C. R. Newman, B. Rhodes, P. A. Chapkin and W. Clegg, J. Organomet. Chem., 2001, 640, 182–196 CrossRef CAS; (e) B. Wang, X. Feng, Y. Huang, H. Liu, X. Cui and Y. Jiang, J. Org. Chem., 2002, 67, 2175–2182 CrossRef CAS PubMed; (f) Z. Zeng, G. Zhao, P. Gao, H. Tang, B. Chen, Z. Zhou and C. Tang, Catal. Commun., 2007, 8, 1443–1446 CrossRef CAS PubMed; (g) M. S. Jeletic, M. T. Jan, I. Ghiviriga, K. A. Abboud and A. S. Veige, Dalton Trans., 2009, 2764–2776 RSC; (h) A. B. Naidu, E. A. Jaseer and G. Sekar, J. Org. Chem., 2009, 74, 3675–3679 CrossRef CAS PubMed; (i) R. J. Lowry, M. T. Jan, K. A. Abboud, I. Ghiviriga and A. S. Veige, Polyhedron, 2010, 29, 553–563 CrossRef CAS PubMed.
- (a) A. Sasaoka, M. Imam Uddin, A. Shimamoto, Y. Ichikawa, M. Shiro and H. Kotsuki, Tetrahedron: Asymmetry, 2006, 17, 2963–2969 CrossRef CAS PubMed; (b) H. R. Cabrera, G. Huelgas, J. M. Hernández Pérez, P. J. Walsh, R. Somanathan and A. de Parrodi, Tetrahedron: Asymmetry, 2015, 26, 163–172 CrossRef PubMed.
- S. Allenmark, U. Skogsberg and L. Thunberg, Tetrahedron: Asymmetry, 2000, 11, 3527–3534 CrossRef CAS.
- (a) M. A. Robjohns, P. Hodge and P. A. Lovell, Polymer, 1997, 38, 3395–3407 CrossRef; (b) S. Zheng and D. Y. Sogah, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2001, 42, 476–477 CAS.
- (a) L. Thunberg and S. Allenmark, Tetrahedron: Asymmetry, 2003, 14, 1317–1322 CrossRef; (b) F. C. Zacconi, L. C. Koll and J. C. Podestá, Tetrahedron: Asymmetry, 2011, 22, 40–46 CrossRef CAS PubMed.
- C. R. Ramanathan and M. Periasamy, Tetrahedron: Asymmetry, 1998, 9, 2651–2656 CrossRef CAS.
- (a) N. Jain and A. V. Bedekar, Tetrahedron: Asymmetry, 2011, 22, 1176–1179 CrossRef CAS PubMed; (b) N. Jain, M. B. Mandal and A. V. Bedekar, Tetrahedron, 2014, 70, 4343–4354 CrossRef CAS PubMed.
- M. J. McKennon, A. I. Meyers, K. Drauz and M. Schwarm, J. Org. Chem., 1993, 58, 3568–3571 CrossRef CAS.
- T. Sakai, Tetrahedron: Asymmetry, 2004, 15, 2749–2756 CrossRef CAS PubMed.
- A. Michaud, C. Lévesque, M. Fila, P. Morin, N. Pelchat and R. Chênevert, Tetrahedron: Asymmetry, 2011, 22, 819–822 CrossRef CAS PubMed.
- (a) K. K.-C. Liu, K. Nozaki and C.-H. Wong, Biocatalysis, 1990, 3, 169–177 CrossRef CAS; (b) S. Akai, T. Naka, Y. Takebo and Y. Kita, Tetrahedron Lett., 1997, 38, 4243–4246 CrossRef CAS.
- H. D. Flack, Acta Crystallogr., Sect. A: Found. Crystallogr., 2009, 65, 371–389 CAS and references cited therein.
- (a) M. Smrcina, M. Lorenc, V. Hanus, P. Sedmera and P. Kocovsky, J. Org. Chem., 1992, 57, 1917–1920 CrossRef CAS; (b) H. Mahmoud, Y. Han, B. M. Segal and L. Cai, Tetrahedron: Asymmetry, 1998, 9, 2035–2042 CrossRef CAS; (c) S. Abela, R. Inauen, D. Spielvogel and C. Moessner, J. Org. Chem., 2012, 77, 4765–4773 CrossRef PubMed; (d) K. Konuki and H. Nagai, Tetrahedron: Asymmetry, 2014, 25, 1581–1589 CrossRef CAS PubMed.
- ESI†.
- (a) V. R. Naidu, M. C. Kim, J.-M. Suk, H.-J. Kim, M. Lee, E. Sim and K.-S. Jeong, Org. Lett., 2008, 10, 5373–5376 CrossRef CAS PubMed; (b) I. Hisaki, N. Shizuki, T. Sasaki, Y. Ito, N. Tohnai and M. Miyata, Cryst. Growth Des., 2010, 10, 5262–5269 CrossRef CAS; (c) K. K. Bisht and E. Suresh, J. Am. Chem. Soc., 2013, 135, 15690–15693 CrossRef CAS PubMed; (d) B. Sureshbabu, R. Venkatachalam and S. Sankararaman, CrystEngComm, 2014, 16, 6098–6106 RSC , and the references cited therein.
- L. Pérez-García and D. B. Amabilino, Chem. Soc. Rev., 2002, 31, 342–356 RSC.
- K. C. Swamy, N. N. Kumar, E. Balaraman and K. V. Kumar, Chem. Rev., 2009, 109, 2551–2651 CrossRef CAS PubMed.
- (a) B. Lal, B. Pramanik, M. S. Manhas and A. K. Bose, Tetrahedron Lett., 1977, 23, 1977–1980 CrossRef; (b) L. E. Overman and D. V. Paone, J. Am. Chem. Soc., 2001, 123, 9465–9467 CrossRef CAS; (c) S. D. Lepore and Y. He, J. Org. Chem., 2003, 68, 8261–8263 CrossRef CAS PubMed.
- W. Kitching, W. Adcock, T. C. Khor and D. Doddrell, J. Org. Chem., 1976, 41, 2055–2058 CrossRef CAS.
- J. S. Meek and R. D. Stack, J. Org. Chem., 1961, 26, 300–302 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of the spectra, X-ray data, TG-DTA. CCDC 1020854, 1020855 and 1054141. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra11626k |
| This journal is © The Royal Society of Chemistry 2015 |