DOI:
10.1039/C5RA11543D
(Paper)
RSC Adv., 2015,
5, 75911-75917
Interaction of bisbenzimidazole-substituted carbazole derivatives with G-quadruplexes and living cells†
Received
16th June 2015
, Accepted 28th August 2015
First published on 1st September 2015
Abstract
G-quadruplex (G4) ligands have potential as chemotherapeutic agents because of the important roles of G4s in regulation of genomic function. Previously, we have developed a fluorescent probe (termed as BPBC) with excellent selectivity to parallel G4s, which possesses a V-shaped bisbenzimidazole-substituted carbazole planar core and two methylpiperazine side arms. Here, we further investigated the interactions of BPBC derivatives with different DNA and living cells. The spectral analysis showed that non-substituted bisbenzimidazole-substituted carbazole (7c) and bisdimethylamino-substituted 7c (7b) exhibited good selectivity to parallel G4s. The binding affinities of BPBC derivatives to parallel G4s were BPBC > 7b ≧ 7c. BPBC and 7b entered living cells and mainly located in the cytoplasma and nucleoli; 7c mainly located in the lysosome. BPBC exhibited the highest cytotoxicity with IC50 around 1 μM. Our results suggest that the bisbenzimidazole-substituted carbazole core is the key factor for selectively binding BPBC derivatives to parallel G4s; the side arms can change their affinity to specific G4s, as well as their interaction with cells. Further optimization of the side arms will provide the opportunity to obtain chemotherapeutic agents targeting specific G4s in cells.
Introduction
G-quadruplex (G4) is a kind of characteristic secondary structure of nucleic acids that contain repetitive tandem guanines. This structure is composed of layers of G-quartets (a planar arrangement of four guanine bases stabilized by eight Hoogsteen hydrogen bonds) and loops. G4-forming sequences are highly prevalent in the human genome,1 especially in promoter regions of oncogenes and telomere ends,2 as well as in particular RNA domains, such as the first introns, 5′- and 3′-untranslated regions and telomeric RNA.3,4 Accumulating evidence has suggested that G4s are involved in many biological processes.5,6 Due to the significant roles in biological systems, G4s have become promising targets for drug discovery,7 therefore, great efforts have been made to design and synthetize specific G4 ligands in the past decade.8–10
Usually G4 ligands consist of a π-conjugated planar core and side chains attached to it. The planar core interacts with G-quartet plane by π–π stacking and the side chains interact with the phosphate backbone, loops or grooves of G4s.11,12 Recently, some ligands with V-shaped planar core have been reported to have good selectivity to G4s over single and double-stranded DNA.13–15 The V-shaped planar core has been considered crucial for G4 binding.16,17 In previous work, we have reported a light up fluorescent probe, BPBC (Fig. 1), which has excellent selectivity to parallel G4s over other G4 structures. BPBC also possesses a V-shaped bisbenzimidazole-substituted carbazole planar core that is slightly larger than the dimension of G-quartet surface.18 However, it is unclear whether this bisbenzimidazole substituted carbazole core is essential for the selectivity of BPBC to parallel G4s. Therefore further comparison of the binding properties of BPBC derivatives will provide deep insights into the molecular design of G4 ligands. Additionally, G4 ligands have the potential to act as the chemotherapeutic anticancer agents through disturbing the functions of G4s in cells. The investigation on the interaction of BPBC and its derivatives with cancer cells will provide helpful information for the drug design based on bisbenzimidazole substituted carbazole derivatives.
 |
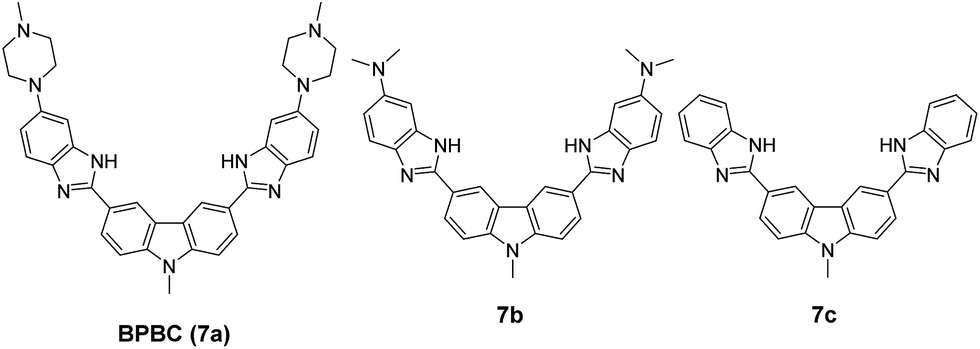
| | Fig. 1 Structures of 7a–c. | |
In this paper, two derivatives of BPBC (7a), 7b and 7c were synthesized. The binding properties of these three compounds to different DNA forms were compared; and their cellular uptake, intracellular localization, and cytotoxicity toward different tumor cells were investigated.
Results and discussion
Design of BPBC derivatives
In order to investigate the contribution of the bisbenzimidazole-substituted carbazole planar core and side arms on the G4 binding of BPBC, two compounds were synthesized (Fig. 1), including the non-substituted bisbenzimidazole-substituted carbazole (7c), a bis-dimethylamino substituted 7c (7b). The synthetic routes of these compounds are shown in the Experimental section.
Absorption and fluorescence spectra of 7b and 7c
BPBC has a tendency to self-aggregate in aqueous medium and to disaggregate in ethanol.18 In order to compare the aggregation behavior of 7b and 7c, their absorption and emission spectra were collected in mixed solvents of ethanol and water. The absorption bands of these compounds were broad in water. Along with the increasing of ethanol, these bands enhanced, became sharp and accompanied with blue shift, which could be considered as the transition of these compounds from aggregated form to monomeric form (see Fig. S1 of the ESI†). Similar with BPBC, 7b and 7c, exhibited very low fluorescence in water, and their fluorescence emission was significantly enhanced accompanying slight blue-shift with the increase of ethanol (see Fig. S1 of the ESI†). In addition, because of the electron-donating ability of amino groups, BPBC and 7b showed longer absorption and emission wavelength than 7c.
Absorption spectra of 7a–c in the presence of different DNA
The interaction between 7a–c and different DNA was investigated by absorption spectra and emission spectra. The used DNA (Table 1) included a single stranded DNA (ss-DNA), a duplex sequence (ds-DNA) formed by ss-DNA and its complementary sequence; four parallel G4 sequences, (EAD,19 c-myc,20,21 Pu22, c-kit2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22); and two antiparallel G4 sequences (thrombin binding aptamer TBA23 and 22AG24).
22); and two antiparallel G4 sequences (thrombin binding aptamer TBA23 and 22AG24).
Table 1 Oligonucleotides used in this study
| Name |
Oligonucleotide sequence (5′ to 3′) |
G4 structure |
| ss-DNA |
CCAGTTCGTAGTAACCC |
Single stranded |
| ds-DNA |
CCAGTTCGTAGTAACCC GGGTTACTACGAACTGG |
Double stranded |
| EAD |
CTGGGTGGGTGGGTGGGA |
Parallel |
| c-Myc |
TGAGGGTGGGGAGGGTGGGGAA |
Parallel |
| Pu22 |
TGAGGGTGGGTAGGGTGGGTAA |
Parallel |
| c-kit2 |
CGGGCGGGCGCGAGGGAGGG |
Parallel |
| TBA |
GGTTGGTGTGGTTGG |
Antiparallel |
| 22AG |
AGGGTTAGGGTTAGGGTTAGGG |
Antiparallel |
As shown in Fig. 2, with the addition of 6-fold excess of different DNA, the absorption bands of 7a–c increased, suggesting that there were certain interactions between DNA and 7a–c. The parallel G4s (EAD, Pu22, c-kit2 and c-Myc) caused significant red-shift or emergence of new peaks in the absorption spectra of 7a–c (Fig. 2 left), but antiparallel G4s (TBA, 22AG), ss-DNAs and ds-DNA caused much smaller change of the absorption spectra of 7a–c (Fig. 2 right), suggesting that 7a–c had much stronger interaction with parallel G4s than with other DNA forms, which was consistent with the results of BPBC reported previously.18 The change extent of absorption spectra (red-shift) of 7a–c upon the addition of different DNA was in the order 7a > 7b ≧ 7c, this order was consistent with the number of amino groups in these compounds, suggesting that there were interactions between side amino groups of 7a/7b and DNA.
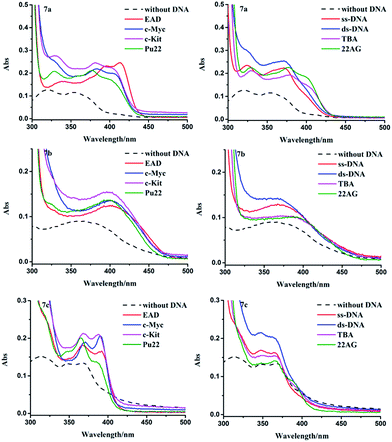 |
| | Fig. 2 Absorption spectra of 7a–c (10 μM) in the presence of different kinds of DNA (60 μM), dash lines represent the spectra of compounds in the absence of DNA. | |
Fluorescence spectra of 7b and 7c in the presence of different DNA
The excitation and emission spectra showed that 7b and 7c exhibited very weak fluorescence in the absence of DNA (Fig. 3). Their fluorescence greatly increased upon the addition of parallel G4s (EAD, Pu22, c-kit2 and c-Myc). But the addition of other forms of DNA only caused weak fluorescence or almost no fluorescence, which agreed well with that of BPBC. This result suggested that 7b and 7c can discriminate between parallel G4s and antiparallel G4s.
 |
| | Fig. 3 Normalized excitation spectra (top) and emission spectra (bottom) of 7b and 7c (1 μM) in the presence of different DNA (6 μM). The excitation spectra of 7b and 7c were collected with λem = 490 and 450 nm. The emission spectra of 7b and 7c were excited with λex = 385 and 375 nm. | |
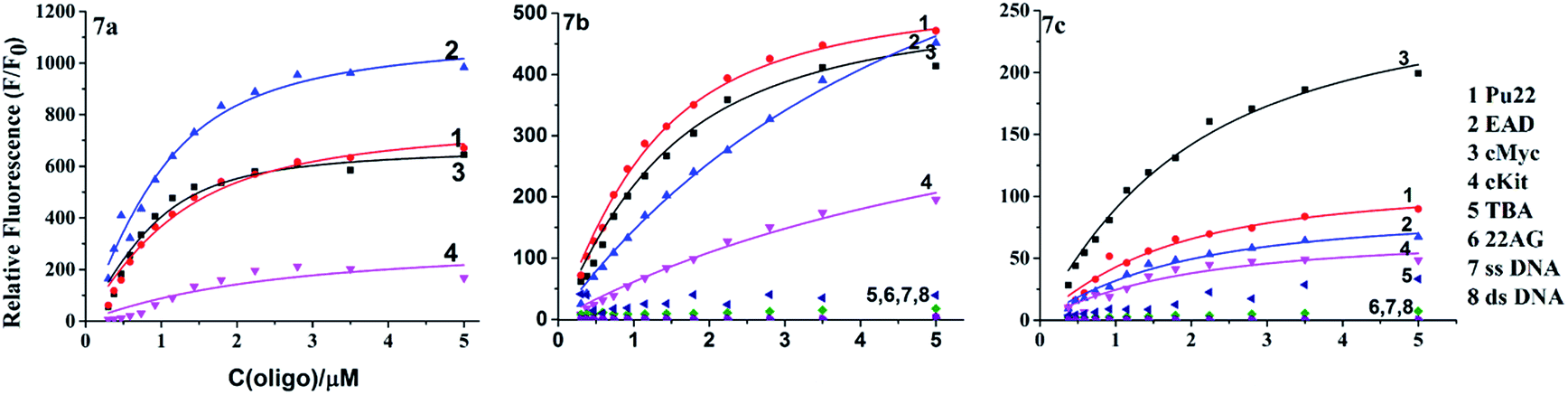
In order to evaluate the binding ability of 7a–c to different types of DNA, the fluorescence titration experiment was performed (Fig. S2–4 of the ESI†). The titration curves (Fig. 4) revealed that 7b and 7c had selectivity to parallel G4s over other forms of DNA, which is consistent with 7a. The titration curves were fitted to an independent-site model25 (eqn (1), materials and methods); the apparent binding constants (Ka) of 7a–c to parallel G4s were calculated as shown in Table 2. 7c showed relatively low binding affinity to all parallel G4s, which may due to its lack of side arms. 7a exhibited much higher binding affinity to c-Myc and higher affinity to EAD and Pu22; 7b exhibited higher affinity to Pu22, c-Myc, and lower affinity to EAD; this difference between 7a and 7c might due to the different interactions of their side arms with the loops or grooves of these G4s.
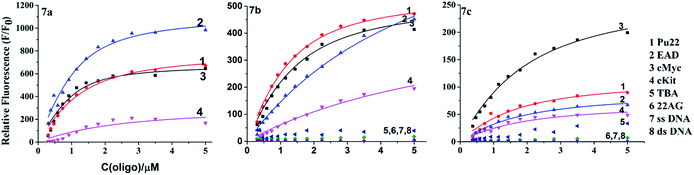 |
| | Fig. 4 Fluorometric titration curves of 7b and 7c (1 μM) with different DNA. | |
Table 2 Apparent binding equilibrium constants (Ka)
| Ka [106 M−1] |
7a |
7b |
7c |
| c-Myc |
3.57 ± 0.11 |
1.19 ± 0.35 |
0.75 ± 0.23 |
| EAD |
2.5 ± 0.09 |
0.21 ± 0.14 |
0.58 ± 0.27 |
| Pu22 |
1.67 ± 0.15 |
1.5 ± 0.02 |
0.68 ± 0.34 |
| c-kit2 |
0.56 ± 0.31 |
0.34 ± 0.81 |
0.51 ± 0.59 |
CD spectroscopy
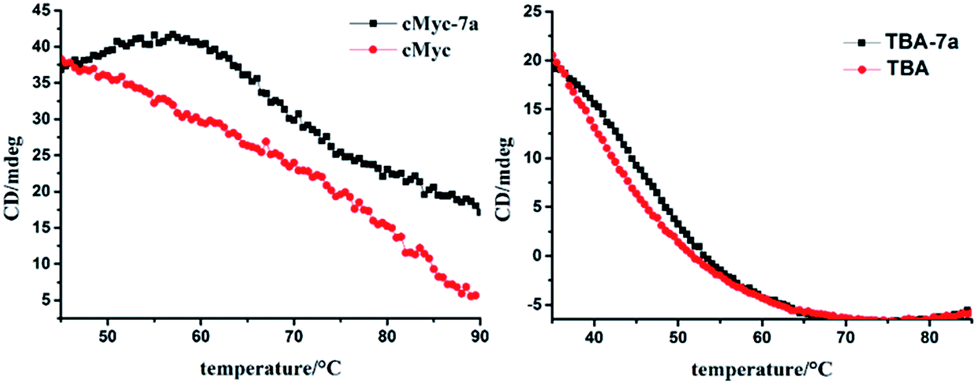
The CD spectra of G4s were measured to study the effect of 7b, 7c on the conformational of G4s. In the absence of the compounds, the CD spectra of parallel G4s (EAD, Pu22) exhibited a characteristic positive peak at 265 nm and a negative peak at 240 nm; antiparallel G4 (TBA and 22AG) showed a characteristic positive peak at 295 nm and a negative band at 260 nm (see Fig. S5 of the ESI†). The addition of 7b or 7c to G4 solutions did not caused the conformational transition; only enhanced the characteristic CD peaks of parallel G4s (EAD and Pu22), and slightly change the characteristic CD peaks of anti-parallel G4s (TBA and 22AG) indicating that the binding of 7b, 7c did not cause significant conformational transition of these G4s, which is similar with 7a. The further CD-based melting study showed that 7a enhanced and maintained the CD signal of the parallel structure of c-myc until the temperature reached over 65 °C and then greatly slowed down the decrease of this CD signal with the temperature raise. However 7a only slightly retarded the decrease of CD signal of the antiparallel structure of TBA (Fig. 5). This result suggests that 7a can selectively stabilize the parallel structure of c-myc, which may imply the potential biological function of 7a.
 |
| | Fig. 5 CD based melting curves of c-myc and TBA in the presence and absence of 7a. The CD signals of c-myc and TBA was collected at 260 nm and 295 nm respectively. | |
All the above spectral results of 7a–c in the absence and presence of different DNA suggest that the bisbenzimidazole-substituted carbazole core structure (7c) is the determinant of the selectivity of 7a–c to parallel G4s. As this core structure can stack on end G-quartet by π–π interaction, and is slightly larger than the dimension of G-quartet plane,18 which makes the side arms on this core approach the groove and loop regions and interact with them. Since the grooves and loops are various depending on the G4 sequences, the different side arms on the core structure may cause different interaction with the different G4s, resulting in different affinity and selectivity to parallel G4s. G4 structures are prevalent in DNA or RNA, a general ligand binding to all G4 structures may disturb the normal function of G4s and result in toxic side-effects as a chemotherapeutic anticancer agents. Our results show the potential to design or screen highly selective ligands for a certain specific parallel G4 by optimizing the side arms on 7c.
Cellular uptake, intracellular localization of 7a, 7b and 7c
G4 ligands have the potential to act as the chemotherapeutic anticancer agents. In order to investigate the potential of bisbenzimidazole-substituted carbazole derivatives as chemotherapeutic anticancer agents in therapy and to obtain the information for drug design, we further compared the cellular uptake, localization and cytotoxicity of these compounds. The confocal imaging showed that all of these three compounds entered living cells (Fig. 6 and S6†). 7a and 7b with amino side arms exhibited higher cellular uptake than 7c based on the fluorescence intensity in cells. The co-staining with Lyso-Tracker Red (lysosome probe) showed that 7a and 7b mainly located in whole cytoplasm and nucleoli, and 7c mainly located in lysosome. These results suggest that the side arms of 7a and 7b increased the cellular uptake and changed the cellular localization. Our previous results have shown that DNA-binding dyes with multiple amino groups mainly locate in nucleus, especially in nucleoli.26 However almost no fluorescence of 7a and 7b was observed in nucleus except in nucleoli, which may due to the low fluorescence emission and weak binding of these compounds to DNA except for parallel G4s. Because G4 formation in gene occurs transitorily when the double-stranded DNA is actively denatured during transcription and replication,27 the fluorescence of 7a and 7b upon binding to G4s might be hardly observed. The fluorescence in cells including in nucleoli may be due to that these compounds located in a low polar environment just like they dissolved in ethanol solution (Fig. S1†).
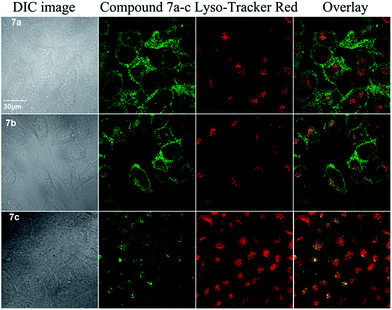 |
| | Fig. 6 Confocal images of MCF-7 cells stained by 7a, 7b or 7c (10 μM) (λex = 405 nm) and Lyso-Tracker Red (λex = 559 nm). | |
Cytotoxicity assay
The cytotoxicity of 7a–c was tested with five cancer cell lines (PC-3, MCF7, HCT-8, A549T, and A549) using CCK-8 assay. After exposed cells to different concentrations of 7a–c for 48 h, the IC50 values of compounds 7a–c were measured. As shown in Table 3, 7a exhibited strong cytotoxicity with IC50 around 1 μM to all the tested cell lines and 7c showed much lower cytotoxicity than 7a. The order of the cytotoxicity was 7a > 7b > 7c, which was consistent with that of the binding ability to G4s and the cellular uptake of these compounds. Although currently we do not know whether the cytotoxicity of these compounds was caused by their G4 binding, the cytotoxicity results suggest that the bisbenzimidazole-substituted carbazole derivatives hold the potential as anticancer reagents. The further investigation will focus on the mechanism of cytotoxicity of BPBC.
Table 3 In vitro cytotoxicity of compounds 7a–c against five human tumor cell lines
| Compound |
IC50 (μM) |
| 7a |
7b |
7c |
| MCF-7 |
1.592 ± 0.04 |
8.193 ± 0.65 |
2.115 ± 0.47 |
| HCT-8 |
0.4457 ± 0.17 |
2.305 ± 0.72 |
62.97 ± 0.24 |
| PC-3 |
1.359 ± 0.08 |
15.371 ± 1.47 |
74.22 ± 2.72 |
| A549 |
0.9984 ± 0.28 |
8.059 ± 0.34 |
17.53 ± 0.48 |
| A549 T |
0.4986 ± 0.06 |
21.93 ± 1.75 |
68.54 ± 1.42 |
Conclusions
In summary, the interaction of three bisbenzimidazole-substituted carbazole derivatives (7a–c) with G-quadruplexes and living cells was investigated. Same as BPBC (7a), bisbenzimidazole-substituted carbazole core of BPBC (7c) and bisdimethylamino-substituted 7c (7b) exhibited good selectivity to parallel G4s. BPBC showed the strongest binding affinity to the tested parallel G4s, and 7b showed better affinity to some of parallel G4s than 7c. Confocal imaging showed that BPBC and 7b could enter into living cells and mainly located in cytoplasma and nucleoli; 7c was mainly found in lysosome. Cytotoxicity assay showed that 7a–c exhibited cytotoxicity to cancer cells, in which 7a exhibited the highest cytotoxicity with IC50 around 1 μM. These results confirmed that bisbenzimidazole-substituted carbazole planar core is the essential element for the specific binding of BPBC derivatives; the side arms can affect their binding ability and selectivity to special G4s and change their cellular uptake, localization and cytotoxicity.
Experimental
Oligonucleotides
All oligonucleotides were purchased from Sunbiotech Co., Ltd (Beijing, China). Oligonucleotides were dissolved in Tris–HCl buffer (10 mM Tris–HCl, 100 mM NaCl, 20 mM KCl and 0.1 mM EDTA, pH = 7.4) except for 22AG. 22AG, was dissolved in (10 mM Tris–HCl, 100 mM NaCl, 0.1 mM EDTA pH = 7.4). The concentration of oligonucleotides was determined based on their absorbance at 260 nm. Oligonucleotides were prepared by heating at 95 °C for 10 min followed with rapidly cooling to 4 °C, and kept at 4 °C overnight.
Materials and reagents
Carbazole, 5-chloro-2-nitroaniline and palladium on activated carbon were obtained from J&K Co., Ltd. Other reagents were purchased from Beijing Chemical Plant (Beijing). Distilled-deionized water was used throughout this work. All chemical reagents were used without further purification. Lyso-Tracker Red kit was bought from Beyotime Institute of Biotechnology (Jiangsu, China). Stock solution of 7a–c (10 mM) was prepared in DMSO.
Cell lines
A549 (non-small cell lung cancer) and MCF-7 (breast cancer) cell lines were purchased from Cell Resource Center of Shanghai Institute for Biological Sciences (Chinese Academy of Sciences, Shanghai, China). PC-3 (prostate cancer) cell lines was purchased from typical culture preservation commission cell bank, Chinese Academy of Sciences (Shanghai, China). A549T (Taxol-resistant A549 subline) cell line was purchased from Shanghai Aiyan Biological Technology Co. Ltd (Shanghai, China). HCT-8 (colon cancer) was purchased from Cell Culture Center of Institute of Basic Medical Sciences (Chinese Academy of Medical Sciences, Beijing, China). Cells were grown in RPMI 1640 Medium (Hyclone) supplemented with 10% fetal bovine serum (FBS, Gibco) at 37 °C in a 5% CO2 humidified atmosphere.
Instruments
1H NMR spectra spectra were recorded at 400 MHz. Low-resolution ESI-mass spectra (MS) were obtained on a LC-MS 2010A (Shimadzu) instrument using standard conditions. MALD-TOF MS were obtained on a Bruker Daltonics flex-Analysis. Fluorescence emission spectra were recorded on a Hitachi F-4500 fluorescence spectrofluorometer (Kyoto, Japan). CD spectra (230–400 nm) were recorded on a Jasco J-815 circular dichroism spectropolarimeter (JASCO Ltd) at a rate of 500 nm min−1 using 400 μL of 1 cm fused quartz cells. Each spectrum was the average of three scans.
 |
| | Scheme 1 Synthetic route of 7a–c. Reagents and conditions: (a) K2CO3, DMF, 110 °C; (b) H2, Pd/C (5%); (c) NaH, DMF, CH3I, r.t; (d) POCl3, DMF, ClCH2CH2Cl, 90 °C; (e) Na2S2O5, EtOH, reflux. | |
Chemistry
General procedures (Scheme 1).
3,6-Diformyl-9-methylcarbazole. To DMF (2.92 g, 40 mmol) at 0 °C, phosphorus oxychloride (6 g, 40 mmol) was added dropwise. The solution was allowed to warm to room temperature, and 9-methylcarbazole (3) (724 mg, 4 mmol) in 1,2-dichloroethane (6 mL) was added. The reaction mixture was heated to 90 °C and kept at this temperature for 24 h. The reaction mixture was poured into water and extracted with chloroform. The chloroform layer was washed with water, and the solvent was removed to yield a deeply colored product, which was purified on a silica gel column using hexane/ethyl acetate (7:3) as eluent, yielding 260 mg (27.4%) of desired product. 1H NMR (400 MHz, CDCl3) δ 10.14 (s, 2H), 8.66 (s, 2H), 8.10 (dd, J = 8.5, 1.4 Hz, 2H), 7.55 (d, J = 8.5 Hz, 2H), 3.97 (s, 3H).
N1,N1-Dimethyl-4-nitrobenzene-1,3-diamine, 5b. 5-Chloro-2-nitro-aniline (25.0 g, 144.9 mmol) was dissolved in DMF (200 mL). Dimethylamine 40 wt% in H2O solution (46 mL) was added into the reaction mixture and then K2CO3 (36 g) was added. The solution was slowly heated to 150 °C and stirred for 12 h. The reaction mixture was poured into excess ice water. The yellow precipitant was filtered and washed with water. The desired product (25.2 g) was obtained in 96% yield. 1H NMR (400 MHz, CDCl3) δ 8.00 (d, J = 9.7 Hz, 1H), 6.19 (s, 2H), 6.13 (dd, J = 9.7, 2.6 Hz, 1H), 5.74 (d, J = 2.6 Hz, 1H), 3.05 (s, 6H).
4 N,N-Dimethylamino-phenylenediamine, 6b. 5b (1 g, 181 g mol−1, 5.5 mmol) was dissolved in 160 mL MeOH/EtOAc (1:1) solvent. 10% Pd/C (110 mg) and NaBH4 (416 mg, 11.0 mmol) was slowly added. The reaction mixture was stirred for about 1 h, after which TLC showed absence of starting material. NH4Cl (400 mg) was added into the reaction mixture and filtered through Celite to remove Pd/C. The filtrate was directly used to further reaction without any purification because this diamino-compound would be oxidatively decomposed during purification steps.
2,2′-(9-Methyl-9H-carbazole-3,6-diyl)bis(N,N-dimethyl-1H-benzo[d]imidazol-5-amine), 7b. To a freshly prepared solution of compound 6b was added 0.5 equiv. of the compound 3, and to this solution was added sodium metabisulfite (Na2S2O5, 1 equiv.) dissolved in a minimum quantity of water. The reaction mixture was refluxed for 7–8 h with stirring, then cooled to room temperature, and filtered through Celite. Ethanol was then removed under reduced pressure to yield the required crude product. This crude product was then purified by column chromatography (EtOAc:MeOH = 2:1) on silica gel to yield the required product yield (20%). 1H NMR (400 MHz, DMSO-d6) δ 9.05 (s, 2H), 8.31 (d, J = 8.6 Hz, 2H), 7.82 (d, J = 8.7 Hz, 2H), 7.49 (d, J = 8.5 Hz, 2H), 6.92–6.81 (m, 4H), 4.03 (s, 3H), 2.97 (s, 12H). 13C NMR (101 MHz, DMSO) δ 154.94, 147.21, 146.15, 138.93, 137.82, 126.55, 123.18, 123.26, 120.47, 115.78, 115.83, 112.78, 105.89, 41.27, 29.60. HRMS (ESI)/HRMS-ESI m/z: calcd for C31H29N7 [M + H]+ 500.25180, found 500.25637.
3,6-Bis(1H-benzo[d]imidazol-2-yl)-9-methyl-9H-carbazole, 7c. To a solution of benzene-1,2-diamine was added 0.5 equiv. of the compound 3, and to this solution was added sodium metabisulfite (Na2S2O5, 1 equiv.) dissolved in a minimum quantity of water. A subsequent synthetic procedure similar to that used for the preparation of compound 7a18 provided compound 7c (yield 25%) as a white powder. 1H NMR (400 MHz, DMSO-d6) δ 12.91 (s, 2H), 9.14 (s, 2H), 8.38 (d, J = 8.5 Hz, 2H), 7.84 (d, J = 8.6 Hz, 2H), 7.84 (d, J = 8.6 Hz, 2H), 7.68 (d, J = 6.2 Hz, 2H), 7.56 (d, J = 6.5 Hz, 2H), 7.22 (s, 2H), 4.02 (s, 3H). 13C NMR (101 MHz, DMSO) δ 152.94, 142.51, 135.43, 125.34, 122.87, 122.59, 122.15, 119.34, 119.06, 117.83, 115.09, 110.47, 109.84, 29.86. HRMS (ESI) calcd for C27H19N5 [M + H]+ 414.16740, found 414.17167.
UV-vis spectra measurement. The absorbance spectra were recorded in Tris–HCl buffer mentioned above on a double beam spectrophotometer UH5300 (HITACHI, Japan).
Fluorimetric titrations. Fluorimetric titrations of the dye 7a–c with the oligonucleotides were performed in Tris–HCl buffer mentioned above. 1 μM 7a–c was mixed with DNA at different concentrations; after standing for 30 min at room temperature in the dark, the fluorescence spectra were collected. The data from the fluorimetric titrations were analyzed according to the independent-site model25 by non-linear fitting to eqn (1),28 in which F0 is the background fluorescence intensity of 7a–c in the absence of DNA, Fmax is the fluorescence intensity upon saturation, and n is the putative number of binding sites on a given DNA. The parameters, Q and A, were found by Levenberg–Marquardt fitting routine in the Origin 8.5 software, whereas n was varied to obtain a better fit.| |
 | (1) |
Circular dichroism spectroscopy. CD spectra were recorded on a JASCO J-815 spectrometer in the wavelength range 200–400 nm using a 10 mm path length quartz cuvette. The scanning speed of the instrument was set to 500 nm min−1, and the response time used was 0.5 s. Each spectrum was the average of three scans. All the strand concentration of oligonucleotides used for measurement was 4 μM. The buffer condition is 10 mM Tris–HCl, 20 mM KCl, 100 mM NaCl and 0.1 mM EDTA.
CD based melting study. Each DNA was dissolved in PBS (10 mM phosphate buffer, 2.7 mM KCl, 137 mM NaCl, pH 7.4) at the final concentration of 4.0 μM and submitted to the annealing procedure (heating to 90 °C and slowly cooling at room temperature). Before each experiment, the samples were equilibrated at 10 °C for 30 min. CD melting curves were obtained by monitoring the variation of CD signals at 295 nm (for TBA) or 260 nm (for c-myc) from 30 to 90 °C. The heating rate was 0.5 °C min−1.
Confocal imaging. Cells were seeded in a 35 mm × 12 mm style cell culture dish (Φ 20 mm glass bottom) and cultured for 24 h in a humided incubator at 37 °C with 5% CO2. After removing the medium, cells were incubated with 1 mL of fresh medium containing compounds (10 μM) and Lyso-Tracker Red (5 μM) was cultured for 2 h. Prior to imaging, cells were washed with phosphate buffered saline (PBS) (pH 7.4) three times, and then imaged under an OLYMPUS FV1000-IX81 confocal microscope (Olympus Corporation, Japan). Fluorescent images were observed using a 100× objective lens and the images were processed using the Olympus FV10-ASW 1.6 viewer software. The excitation wavelengths were 405 nm and fluorescence signals were collected from 425–475 nm. The Lyso-Tracker Red were excited at 559 nm, and monitored at 610–660 nm.
In vitro cytotoxicity studies. The cytotoxicity of compounds 7a–c was evaluated by Cell Counting Kit-8 (CCK-8) kit (Dojindo Laboratories, Kumamoto, Japan). In brief, MCF7, HCT-8, PC-3, A549 or A549 T cells (5 × 103 well) were seeded into 96-well plates and incubated at 37 °C for 18 h. Cells were then treated with the indicated concentrations of compounds, respectively. The plates were incubated at 37 °C for an additional 48 h. Then the medium was replaced with 100 mL of fresh medium (without FBS and penicillin/streptomycin) containing 10 mL of CCK-8 reagent. After incubation at 37 °C/5% CO2 for 1 h, the absorbance at 450 nm was measured on a SpectraMax M5 (Molecular Devices, CA, USA). The cell viability rate (VR) was calculated according to the eqn (2):| |
 | (2) |
where A is the absorbance of the experimental group, As is the absorbance of the control group and A0 is the absorbance of the blank group (no cells).
Acknowledgements
We gratefully acknowledge the financial support from Grant 973 Program (2011CB935800, 2011CB911000 and 2013CB933700), NSF of China (21275149, 21375135, 21205124 and 21321003) and NSF of Guangxi Province of China (2013GXNSFBA019158).
Notes and references
- A. K. Todd, M. Johnston and S. Neidle, Nucleic Acids Res., 2005, 33, 2901–2907 CrossRef CAS PubMed.
- J. L. Huppert and S. Balasubramanian, Nucleic Acids Res., 2007, 35, 406–413 CrossRef CAS PubMed.
- J. Eddy and N. Maizels, Nucleic Acids Res., 2008, 36, 1321–1333 CrossRef CAS PubMed.
- J. L. Huppert, A. Bugaut, S. Kumari and S. Balasubramanian, Nucleic Acids Res., 2008, 36, 6260–6268 CrossRef CAS PubMed.
- T. A. Brooks, S. Kendrick and L. Hurley, FEBS J., 2010, 277, 3459–3469 CrossRef CAS PubMed.
- P. Agarwala, S. Pandey and S. Maiti, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 3503–3510 CrossRef CAS PubMed.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef CAS PubMed.
- T. M. Ou, Y. J. Lu, J. H. Tan, Z. S. Huang, K. Y. Wong and L. Q. Gu, Chemmedchem, 2008, 3, 690–713 CrossRef CAS PubMed.
- S. N. Georgiades, N. H. Abd Karim, K. Suntharalingam and R. Vilar, Angew. Chem., Int. Ed., 2010, 49, 4020–4034 CrossRef CAS PubMed.
- J. H. Guo, L. N. Zhu, D. M. Kong and H. X. Shen, Talanta, 2009, 80, 607–613 CrossRef CAS PubMed.
- A. K. Jain and S. Bhattacharya, Bioconjugate Chem., 2011, 22, 2355–2368 CrossRef CAS PubMed.
- L. N. Zhu, S. J. Zhao, B. Wu, X. Z. Li and D. M. Kong, PLoS One, 2012, 7, e35586 CAS.
- B. Jin, X. Zhang, W. Zheng, X. Liu, J. Zhou, N. Zhang, F. Wang and D. Shangguan, Anal. Chem., 2014, 86, 7063–7070 CrossRef CAS PubMed.
- C. C. Chang, J. Y. Wu and T. C. Chang, J. Chin. Chem. Soc., 2003, 50, 185–188 CrossRef CAS PubMed.
- C. C. Chang, I. C. Kuo, I. F. Ling, C. T. Chen, H. C. Chen, P. J. Lou, J. J. Lin and T. C. Chang, Anal. Chem., 2004, 76, 4490–4494 CrossRef CAS PubMed.
- A. De Cian and J. L. Mergny, Nucleic Acids Res., 2007, 35, 2483–2493 CrossRef CAS PubMed.
- S. Bhattacharya, P. Chaudhuri, A. K. Jain and A. Paul, Bioconjugate Chem., 2010, 21, 1148–1159 CrossRef CAS PubMed.
- B. Jin, X. Zhang, W. Zheng, X. J. Liu, C. Qi, F. Y. Wang and D. H. Shangguan, Anal. Chem., 2014, 86, 943–952 CrossRef CAS PubMed.
- X. H. Cheng, X. J. Liu, T. Bing, R. Zhao, S. X. Xiong and D. H. Shangguan, Biopolymers, 2009, 91, 874–883 CrossRef CAS PubMed.
- A. T. Phan, V. Kuryavyi, H. Y. Gaw and D. J. Patel, Nat. Chem. Biol., 2005, 1, 167–173 CrossRef CAS PubMed.
- J. X. Dai, M. Carver, L. H. Hurley and D. Z. Yang, J. Am. Chem. Soc., 2011, 133, 17673–17680 CrossRef CAS PubMed.
- S. T. D. Hsu, P. Varnai, A. Bugaut, A. P. Reszka, S. Neidle and S. Balasubramanian, J. Am. Chem. Soc., 2009, 131, 13399–13409 CrossRef CAS PubMed.
- L. Martino, A. Virno, A. Randazzo, A. Virgilio, V. Esposito, C. Giancola, M. Bucci, G. Cirino and L. Mayol, Nucleic Acids Res., 2006, 34, 6653–6662 CrossRef CAS PubMed.
- A. Ambrus, D. Chen, J. X. Dai, T. Bialis, R. A. Jones and D. Z. Yang, Nucleic Acids Res., 2006, 34, 2723–2735 CrossRef CAS PubMed.
- F. H. Stootman, D. M. Fisher, A. Rodger and J. R. Aldrich-Wright, Analyst, 2006, 131, 1145–1151 RSC.
- J. Zhou, A. Chang, L. L. Wang, Y. Liu, X. J. Liu and D. H. Shangguan, Org. Biomol. Chem., 2014, 12, 9207–9215 CAS.
- N. Maizels, Nat. Struct. Mol. Biol., 2006, 13, 1055–1059 CAS.
- X. Xie, B. Choi, E. Largy, R. Guillot, A. Granzhan and M. P. Teulade-Fichou, Chem.–Eur. J., 2013, 19, 1214–1226 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further details are given in Fig. S1–6. For MS and 1H NMR spectra. See DOI: 10.1039/c5ra11543d |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.