
Physically crosslinked poly(vinyl alcohol)–carrageenan composite hydrogels: pore structure stability and cell adhesive ability
Abstract
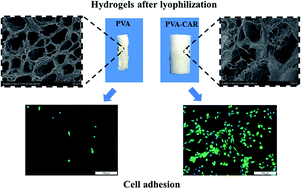
Poly(vinyl alcohol) (PVA) hydrogels have gained comprehensive attention in the biomedical area. However, their resistance to cell adhesion is a drawback for applications such as tissue engineering. Besides, the controllability of the porous structure of PVA-based hydrogels during lyophilization needs to be further improved. Herein, we prepared PVA–carrageenan (CAR) composite hydrogels as tissue engineering scaffolds using the freeze–thaw technique. The hydrogels were found to possess deformation resistance, preserving their shape during the lyophilization process without shrinkage. Besides, ATDC5 cells showed good adherence and proliferation activity on the composite hydrogels. In addition, the PVA–CAR composite hydrogels possess good hemocompatibility and did not cause any adverse effects in the inflammatory response from RAW 264.7 macrophage cells. Overall, the results obtained indicate that the PVA–CAR composite hydrogels show potential applications in the field of tissue engineering based on their good structural stability, excellent biocompatibility and mild fabrication process.
 Please wait while we load your content...
Please wait while we load your content...

