DOI:
10.1039/C5RA16299H
(Paper)
RSC Adv., 2015,
5, 78172-78179
Surface modification of polysulfonamide fiber treated by air plasma
Received
13th August 2015
, Accepted 3rd September 2015
First published on 3rd September 2015
Abstract
The surface of polysulfonamide (PSA) fiber was modified by air plasma to improve the wettability and interfacial bonding performance of the fiber. The optical contact angle and the single fiber pull-out method were used to determine the wettability and interfacial bonding performance of the PSA fibers before and after surface modification. The surface morphology and chemical composition of the fiber were evaluated by field emission scanning electron microscopy (FE-SEM), atomic force microscopy (AFM), and X-ray photoelectron spectrometry (XPS). Results showed that after air plasma treatment, the water absorption time decreased from >400 s to approximately 0 s and the wettability of the PSA fiber increased sharply. The increase in processing time also improved the PSA fiber wettability. After air plasma treatment for 3 min, the interfacial shear strength reached a peak value of 11.19 MPa, an increase of 56.94% compared with the untreated specimen. FE-SEM and AFM observations confirmed that the PSA fiber surface roughened with a prolonged treatment duration. XPS analysis showed that the O/C atomic ratio on the PSA fiber surface can be increased from 26.93% to 47.21% after 3 min of treatment. At such conditions, the oxygen-containing polar groups reached the highest levels, which enhanced the wettability and interfacial bonding performance considerably. Furthermore, the single fiber tensile strength test showed that the PSA fiber breaking strength exhibited only slight reductions after a short air plasma treatment time.
Introduction
Polysulfonamide (PSA) is a high-temperature resistant fiber with independent intellectual property rights owned by the Chinese. PSA fiber has an initial decomposition temperature higher than 400 °C and can be used for a long time at temperatures up to 250 °C. Fig. 1 illustrates the chemical structure of PSA fibers. Due to the additional sulfone group (–SO2–) structure in its molecular main chain and the existence of the conjugated aromatic rings, the electron density of the amide is significantly reduced, and the PSA fiber cannot be destroyed at high temperature.1 PSA fibers possess numerous desirable properties, such as high tensile strength and modulus, flame resistance, chemical corrosion, resistance to high temperature, electrical insulating properties, and anti-electromagnetic radiation. Therefore, this fiber can be applied in several areas, such as metallurgy, fire control, military projects, and space navigation. More importantly, PSA plays an important role as a reinforcing component in composite materials.2 However, PSA fiber shows poor bonding properties between the fiber and the resin matrix because of its smooth surface, few polar groups, low surface energy, and poor wettability.2,3 Interfacial fractures often appear during use, thereby affecting the performance of the PSA fiber composites. Hence, modification of the PSA fibers is necessary to improve their surface properties. Traditional chemical treatments often use water and/or organic solvents, but the disposal of the drained water and the recovery of the organic solvent cause environmental pollution and wasteful use of resources.4,5 Flame treatment, corona discharge, and plasma have been suggested as suitable approaches to enhance polymer adhesion, although the most effective and feasible method is still under debate.6–8 However, researchers generally agree that the effective duration is the longest after plasma treatment.9 Plasma surface modification technology is a non-aqueous, low energy consuming, and environmentally friendly processing method.10–13 Wang et al.2 showed that the water absorption time of PSA yarns decreases from more than 300 s to about 5 s after helium-plasma treatment. Improving the wettability, adhesion, and surface properties of fiber or polymer materials with plasma treatment has been successfully applied in research for the past few years.14–23 Up till now, there is no related report on surface modification of PSA fiber treated by air plasma.
 |
| | Fig. 1 The chemical structure of PSA fiber. | |
The specimen preparation for a single-fiber pull-out test described in the literature is complicated and difficult to control.24–26 Thus, a new method was developed to simplify the experimental operations and to reduce the corresponding experimental errors accompanying the use of a self-created card method. The tested interfacial shear strength (IFSS) deviated only by ≤4% from that of the method described in the literature.25,26 The present study focuses on the influences of the air plasma treatment on the wettability and the interfacial properties of PSA fibers. Field emission scanning electron microscopy (FE-SEM), atomic force microscopy (AFM), and X-ray photoelectron spectrometry (XPS) were conducted to determine the surface changes in morphology and chemical composition after air plasma.
Experimental
Materials
PSA fibers with a diameter of 15 μm and a density of 1.42 g cm−3 were provided by Shanghai Tanlon Fiber Co., Ltd in China. Acetone was purchased from Tianjin Chemical Reagent Factory (China). Epoxy resin (WSR6101, bisphenol-A type epoxy) and curing agent (651) were supplied by BlueStar new chemical materials Co., Ltd in China.
Sample preparations
PSA fibers were washed in acetone for 30 min to remove the finishes and surface contaminants. The fibers were then thoroughly rinsed with distilled water and dried under vacuum for more than 12 h to eliminate the remaining acetone completely. After washing, the fibers were equally divided into six portions and then immediately placed in a clean plastic bag.
Plasma treatments
The plasma treatment system (PR-3) was provided by Beijing Creation Weina Technology Co., Ltd (China) with the following experimental conditions: a plasma power of 100 W and a pressure of 40 Pa. The PSA fibers were placed in the glass sample stage and were treated by air plasma for different durations (1, 2, 3, 4, and 5 min). Subsequently, the fibers were placed in a clean plastic bag to minimize potential contamination.
Preparation of the PSA fiber composite material
Epoxy resin (WSR6101) and curing agent (651) with a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were dissolved in acetone and were subsequently churned into a homogeneous adhesive solution. A single PSA fiber was pasted onto the center of a paper sample card, as shown in Fig. 2, which we personally constructed. A trace adhesive solution measured with a syringe needle was dropped onto the PSA fiber. During dropping, a uniform distribution of adhesive droplets was ensured and diameters of less than 1 mm were maintained. The specimens were cured at 90 °C for 2 h. An industrial microscope (BX51TRF; Olympus) was employed to measure the diameter of each fiber, the diameter of the resin beads, and the embedded length.
:1 were dissolved in acetone and were subsequently churned into a homogeneous adhesive solution. A single PSA fiber was pasted onto the center of a paper sample card, as shown in Fig. 2, which we personally constructed. A trace adhesive solution measured with a syringe needle was dropped onto the PSA fiber. During dropping, a uniform distribution of adhesive droplets was ensured and diameters of less than 1 mm were maintained. The specimens were cured at 90 °C for 2 h. An industrial microscope (BX51TRF; Olympus) was employed to measure the diameter of each fiber, the diameter of the resin beads, and the embedded length.
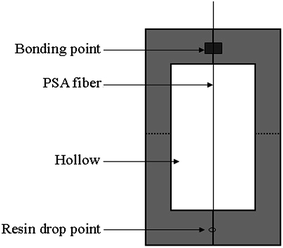 |
| | Fig. 2 Sample card. | |
FE-SEM observation
A field emission scanning electron microscope (FE-SEM, S4800; Hitachi, Japan) was used to observe the surface morphology of the fibers before and after air plasma treatment. Prior to the FE-SEM observation, the fibers were coated with a thin layer of gold to improve their surface conductivity. FE-SEM images were taken at 8000× magnification.
AFM observation
An atomic force microscope (CSPM5500; BenYuan) was employed to characterize the surface morphology of the PSA fibers. A tapping mode was used to scan the fiber surface. AFM images of the PSA fiber were obtained with a scan area of 8 μm × 8 μm. The surface roughness of the PSA fibers before and after air plasma treatment was characterized and compared by the root mean square roughness (Sq) and the roughness average (Sa), which were calculated using the CSPM Imager software.
XPS analysis
An X-ray photoelectron spectrometer (K-Alpha; ThermoFisher, USA) was employed to analyze the surface chemical composition of the fibers before and after air plasma treatment. The test system was equipped with a Mg Kα X-ray source (1253.6 eV), and the analysis was performed under ultra-high vacuum conditions (10−7 Pa to 10−8 Pa).
Wettability measurement
The PSA fiber wettability was evaluated by measuring the optical contact angle and water absorption time according to the sessile drop technique.2,23,31 A contact angle analyzer (JC2000D2; Shanghai) was used to measure the optical contact angle and the water absorption time at the condition of 20 °C and 65% relative humidity, by taking 0.02 g of carded straight PSA fiber and then attaching it to a plastic sheet, maintained with the same distance. A distilled water droplet of 5 μl was placed on the fiber surface by using a microliter syringe. The initial contact angle was recorded as soon as the water droplet was on the PSA fiber surface. The time of complete droplet absorption into the fibers was recorded as the water absorption time. A total of 20 measurements were performed for each sample.
Adhesion measurement
The adhesion of the fiber and epoxy resin was investigated by conducting a pull-out test, and their strengths were determined with a universal testing machine (Instron 3369; USA) at a displacement of 1 mm min−1. The fiber sample card was folded along the central vertical line and then clamped in the upper and lower collets. The card was cut along the dotted line, and the influence of the card strength was eliminated. Twenty measurements were performed for each sample. The formula is expressed as follows:| |
 | (1) |
where IFSS is the interfacial shear strength, Pmax is the peak load, L is the imbedded length, r is the radius of the fiber, A is the cross-sectional area of the fiber, and n is defined as:| |
 | (2) |
where Em and Ef are the Young’s moduli of the matrix (1.3 GPa) and the fiber (7.45 GPa), respectively, Vm is the Poisson’s ratio of the matrix (0.39), and R is the radius of the resin beads.
Breaking strength measurement
A single fiber electronic tensile strength tester (YM-06A; YuanMore Instrument Co., Ltd, China) was employed to measure the breaking strength of the PSA fibers. The gauge length, drawing speed, and pre-tension for the tensile tests were 20 mm, 20 mm min−1, and 0.2 cN, respectively. Thirty measurements were performed for each sample.
Statistical analysis
Tukey’s test and a one-way ANOVA were used to compare the breaking strength, water absorption time and IFSS value.27 P-values of less than 0.05 were considered significant.
Results and discussion
Surface morphology and roughness
FE-SEM was conducted to observe the surface morphologies of the PSA fiber before and after air plasma treatment (Fig. 3). Compared with the untreated fiber, the surface of the original fiber was relatively smooth and then became uneven and clearly different after air plasma treatment. In addition, the etching phenomenon became more evident with longer treatment times. This phenomenon results from the oxidative reactions induced by air plasma processing.28,29
 |
| | Fig. 3 FE-SEM micrographs of the PSA fiber surface before and after air plasma treatment: (a) untreated, (b) 1 min, (c) 3 min and (d) 5 min. | |
AFM was performed to analyze the change of the PSA fiber surface roughness before and after air plasma treatment. Fig. 4 shows that air plasma treatment can change the fiber surface morphology on a microscopic scale. Table 1 illustrates the roughness of PSA samples obtained from the AFM images.
 |
| | Fig. 4 AFM images of the PSA fiber surface before and after air plasma treatment: (a) untreated, (b) 1 min, (c) 3 min and (d) 5 min. | |
Table 1 Root mean square roughness (Sq) and roughness average (Sa) of PSA fibers
| Fiber sample |
Sq (nm) |
Sa (nm) |
| Untreated |
20.3 |
15.0 |
| Air plasma treated 1 min |
42.6 |
33.9 |
| Air plasma treated 3 min |
46.4 |
36.5 |
| Air plasma treated 5 min |
50.1 |
41.2 |
The surface of the original fiber (Fig. 4a) appeared relatively smooth. The root mean square roughness (Sq) and roughness average (Sa) of the untreated sample were 20.3 and 15.0 nm, respectively. At 1 min treatment time (Fig. 4b), the fiber surface exhibited some micro-pits and the surface roughness was evidently increased. When the treatment time was increased to 3 min (Fig. 4c), numerous pits were found on the fiber surface. In this sample, Sq and Sa increased further to 46.4 and 36.5 nm, respectively. When the treatment time was further increased to 5 min (Fig. 4d), the surface roughness persistently increased, accompanied by the appearance of many large bulges with irregular shapes. These results correlate well with the FE-SEM analysis.
The increase in the surface roughness is beneficial for improving the wettability and interfacial bonding strength of the fibers. However, the PSA fiber surface produces more convex precipitates and a weak boundary effect with long treatment duration. The interfacial bonding strength of the fiber composites is also reduced.
Surface chemical composition
Table 2 shows that the content of C, O, and N on the PSA fiber surface are closely related to the sample treatment time. The untreated fiber had an oxygen content of 19.5%, whereas the samples after 1, 3, and 5 min air plasma treatment exhibited oxygen content of 23.6%, 28.8%, and 23.1%, respectively. Similarly, the O/C atomic ratio initially increased from 26.93% to 47.21% and then decreased. The change of the N and N/C atomic ratios displayed the same trend as that of the O and O/C atomic ratios, but the trend was not evident. These results were attributed to the introduction of oxygen and nitrogen to the fiber surface after oxidation or nitriding during the air plasma modification. The incorporation of these polar functional groups can be partly responsible for the considerable enhancement of the wettability of the PSA fibers treated by air plasma. However, when the fibers were treated by air plasma for 5 min, the content of N and O decreased because of the dominant air plasma etching effects.
Table 2 Relative chemical composition and atomic ratios determined by XPS for the PSA fiber surface before and after air plasma treatment
| Sample |
Chemical composition (%) |
Atomic ratio (%) |
| C1s |
O1s |
N1s |
S2p |
O/C |
N/C |
(O + N)/C |
| Untreated |
72.4 |
19.5 |
5.8 |
2.3 |
26.93 |
8.01 |
34.94 |
| Air plasma treated 1 min |
67.5 |
23.6 |
6.5 |
2.4 |
34.96 |
9.63 |
44.59 |
| Air plasma treated 3 min |
61.0 |
28.8 |
7.7 |
2.5 |
47.21 |
12.62 |
59.83 |
| Air plasma treated 5 min |
67.1 |
23.1 |
7.3 |
2.5 |
34.43 |
10.88 |
45.31 |
XPSPEAK, a peak split software, was used to analyze the changes in the content of the chemical groups on the PSA fiber surface before and after plasma treatment with the corresponding spectrum of obtained C1s-split specific groups. Table 3 and Fig. 5 present the values of the bonding energy in the functional groups containing carbon, such as –C–C–, –C–N–/–C–O–, –CONH–, and –O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–OH, which were 285.0, 286.3, 287.8, and 289.0 eV, respectively. Compared with the untreated fiber, the –C–C– group concentration decreased dramatically from 54.12% to 30.98% after air plasma treatment for 3 min. The concentrations of –C–N–/–C–O–, –CONH–, and –OC–OH remarkably increased from 28.68%, 14.89%, and 2.31% to 43.04%, 17.95%, and 8.03%, respectively, reaching the highest levels in this study. In the PSA fiber treated with 5 min air plasma, the –C–C– group showed a significant increase and the aforementioned oxygen-containing polar groups decreased sharply to 35.12%, 17.01%, and 7.02%, respectively, compared with the 3 min treatment. This observation resulted from the ablation/etching of the extended treatment time, which reversed the surface functionalization. Notably, a C–N contribution must be present in the C1s spectrum at approximately the same position as C–O on 286.3 eV and –CONH– on 287.8 eV; however, these contributions are relatively small.30 XPS results also indicate that the oxygen and polar functional groups incorporated into the PSA fiber surface can achieve preferable levels at an appropriate air plasma treatment time, which plays an important role in the improvement of the PSA fiber surface wettability and bonding strength between the fiber and resin.
C–OH, which were 285.0, 286.3, 287.8, and 289.0 eV, respectively. Compared with the untreated fiber, the –C–C– group concentration decreased dramatically from 54.12% to 30.98% after air plasma treatment for 3 min. The concentrations of –C–N–/–C–O–, –CONH–, and –OC–OH remarkably increased from 28.68%, 14.89%, and 2.31% to 43.04%, 17.95%, and 8.03%, respectively, reaching the highest levels in this study. In the PSA fiber treated with 5 min air plasma, the –C–C– group showed a significant increase and the aforementioned oxygen-containing polar groups decreased sharply to 35.12%, 17.01%, and 7.02%, respectively, compared with the 3 min treatment. This observation resulted from the ablation/etching of the extended treatment time, which reversed the surface functionalization. Notably, a C–N contribution must be present in the C1s spectrum at approximately the same position as C–O on 286.3 eV and –CONH– on 287.8 eV; however, these contributions are relatively small.30 XPS results also indicate that the oxygen and polar functional groups incorporated into the PSA fiber surface can achieve preferable levels at an appropriate air plasma treatment time, which plays an important role in the improvement of the PSA fiber surface wettability and bonding strength between the fiber and resin.
Table 3 Results of the deconvolution of C1s peaks for the PSA fiber surface before and after air plasma treatment
| Sample |
Carbon groups concentration (%) |
| –C–C– |
–C–N–/–C–O– |
–CONH– |
–OC–OH |
| Untreated |
54.12 |
28.68 |
14.89 |
2.31 |
| Air plasma treated 1 min |
47.96 |
31.87 |
16.13 |
4.04 |
| Air plasma treated 3 min |
30.98 |
43.04 |
17.95 |
8.03 |
| Air plasma treated 5 min |
40.85 |
35.12 |
17.01 |
7.02 |
 |
| | Fig. 5 C1s XPS spectra of the PSA fiber surface before and after air plasma treatment: (a) untreated, (b) 1 min, (c) 3 min and (d) 5 min. | |
Wettability of PSA fiber
Fig. 6 shows that the water absorption time dropped from >400 s to approximately 0 s. At the start of the air plasma treatment, the water absorption time of the PSA fiber declined significantly and then slowly after 3 min. The time dropped within 1.3 s after 3 min of treatment and almost approached zero after 5 min. The effect of evaporation can be considered to be negligible because the water-absorption time of the PSA fiber after air-plasma treatment was <7 s in this study.31 Fig. 7 shows the effect of the air-plasma treatment time on the contact angle of the PSA fiber. The contact angles of the untreated PSA fiber and fiber treated for 3 min were 101° and 44°, respectively. The contact angle of the PSA fiber declined slowly after more than 3 min of treatment, whereas the contact angle was 33° after 5 min of treatment. These results correlated well with the water-absorption time test. Water droplet images showed that the perceptible reduction in the solid–liquid contact area was accompanied by a steep decrease in the volume after air-plasma treatment.
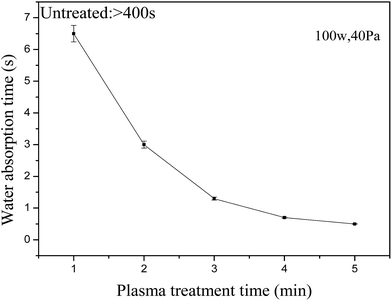 |
| | Fig. 6 Effect of the air plasma treatment time on the water absorption times of PSA fiber. | |
 |
| | Fig. 7 Effect of the air plasma treatment time on the contact angles of PSA fiber. | |
These results can be explained by the increase in the surface roughness of the PSA fiber by the air plasma, as evidenced by the AFM images, and Wenzel and Cassie theories.32 It can also be attributed to the introduction of some new polar groups onto the fiber surface as shown by the XPS analysis. These reasons improved the PSA fiber surface wettability. After air plasma treatment, the wettability of the PSA fiber was apparently enhanced, which facilitates the spreading of the resin on the fiber surface, and increases the interfacial adhesion of the composite materials.
Adhesion of PSA fiber
Fig. 8 shows that the IFSS value of the PSA fiber increased after air plasma treatment. The IFSS values of the untreated PSA fiber and the fiber treated for 1 min were 7.13 and 9.25 MPa, respectively. After 3 min, the IFSS reached a peak value of 11.19 MPa, an increase of 56.94% compared with the untreated sample. The increased surface roughness and wettability of the PSA fiber resulted in an increased physical adhesion between the fiber and resin. After plasma treatment, the number of polar groups on the fiber surface increased. This result indicates that the chemical adhesion between the fiber and resin was enhanced. Physical and chemical adhesions determined the boundary strength of the composite material of the fiber and resin. After more than 3 min of treatment, the IFSS value began to drop because the long plasma treatment time decreased the fiber breaking strength. Consequently, the fiber surface produced some convex precipitates and debris, and the polar groups on the fiber surface began to diminish, which produced a weak boundary effect with the interfacial cohesion of the reduced fiber. This result is consistent with those obtained from the FE-SEM, AFM, and XPS tests.
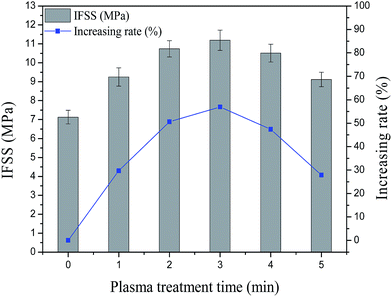 |
| | Fig. 8 Effect of the air plasma treatment time on the IFSS values of PSA fiber. | |
Breaking strength of PSA fiber
As shown in Fig. 9, a short air plasma treatment caused minor effects on the breaking strength of the PSA fiber, and the breaking strength after 3 min of treatment was reduced by 2.6%. After more than 3 min of treatment, the breaking strength of the PSA fiber declined rapidly, whereas after 5 min of treatment, the strength was reduced by 8.1%, because with the increasing air plasma treatment time, the degree of plasma etching increases, as well as the stress concentration in the flaws which are introduced to the fiber surface by air plasma etching.4 Air plasma technology was used to enhance the fiber surface properties. Mechanical performance of the fiber should also be considered.
 |
| | Fig. 9 Effect of the air plasma treatment time on the breaking strength of PSA fiber. | |
Conclusions
A short treatment time of air plasma can enhance the PSA fiber surface wettability and IFSS, which is attributed not only to the production of polar groups at the PSA fiber surface but also to the observed surface roughness modification.
FE-SEM and AFM observations confirmed that with increasing air plasma treatment time, the surface roughness of the PSA fiber and the degree of plasma etching increased. After the air plasma treatment, the polar groups created at the PSA fiber surface, such as –C–O–, –CONH–, and –OC–OH, were analyzed by XPS and showed prominent increases compared with those of the untreated sample. After air plasma treatment for 3 min, the oxygen-containing polar groups reached the highest levels in this study.
With the air plasma treatment conditions at 40 Pa, 100 W, and 3 min, the breaking strength of the PSA fiber dropped by 2.6%, the IFSS value reached the peak value of 11.19 MPa, and the water absorption time decreased from >400 s to 1.3 s. However, after more than 3 min of treatment, the breaking strength of the PSA fiber declined rapidly, and the IFSS value began to drop. Therefore, an optimum air plasma treatment time of 3 min was determined.
Acknowledgements
This work was supported by Tianjin Science and Technology Plan Projects [No. 12TXGCCX01900].
Notes and references
- H. M. Li, Y. Zhu, B. Xu, C. X. Wu, J. X. Zhao and M. X. Dai, J. Appl. Polym. Sci., 2013, 127, 342 CrossRef CAS PubMed.
- C. X. Wang, J. C. Lv, D. W. Gao, G. L. Liu, L. M. Jin and J. H. Liu, Fibers Polym., 2013, 14, 1478 CrossRef CAS.
- L. N. Ndlovu, Q. Siddiqui, E. Omollo and C. W. Yu, Text. Res. J., 2015, 85, 262 CrossRef CAS PubMed.
- C. X. Jia, P. Chen, W. Liu, B. Li and Q. Wang, Appl. Surf. Sci., 2011, 257, 4165 CrossRef CAS PubMed.
- Z. Q. Gao, J. Sun, S. J. Peng, L. Yao and Y. P. Qiu, J. Appl. Polym. Sci., 2011, 120, 2201 CrossRef CAS PubMed.
- I. Novák, V. Pollák and I. Chodák, Plasma Processes Polym., 2006, 3, 355 CrossRef PubMed.
- S. Farris, S. Pozzoli, P. Biagioni, L. Duó and S. Mancinelli, Polymer, 2010, 51, 3591 CrossRef CAS PubMed.
- M. Strobel, V. Jones, C. S. Lyons, M. Ulsh, M. J. Kushner, R. Dorai and M. C. Branch, Plasmas Polym., 2003, 8, 61 CrossRef CAS.
- M. Strobel, M. J. Walzak, J. M. Hill, A. Lin, E. Karbashewski and C. S. Lyons, J. Adhes. Sci. Technol., 1995, 9, 365 CrossRef CAS PubMed.
- A. Demir, Fibers Polym., 2010, 11, 580 CrossRef.
- M. J. Perez-Roldan, D. Debarnot and F. Poncin-Epaillard, RSC Adv., 2014, 4, 64006 RSC.
- S. Inbakumar, R. Morent, N. D. Geyter, T. Desmet, A. Anukaliani, P. Dubruel and C. Leys, Cellulose, 2010, 17, 417 CrossRef CAS.
- C. C. Lai and C. T. Lo, RSC Adv., 2015, 5, 38868 RSC.
- N. D. Boscher, V. Vache, P. Carminati, P. Grysan and P. Choquet, J. Mater. Chem. A, 2014, 2, 5744 CAS.
- S. Ratnapandian, L. J. Wang, S. M. Fergusson and M. Naebe, J. Fiber Bioeng. Informat., 2011, 4, 267 CrossRef.
- M. Golda-Cępa, K. Engvall and A. Kotarba, RSC Adv., 2015, 5, 48816 RSC.
- H. L. Xu, S. J. Peng, C. X. Wang, L. Yao, J. Sun, F. Ji and Y. P. Qiu, J. Appl. Polym. Sci., 2009, 113, 3687 CrossRef CAS PubMed.
- D. Gogoi, A. J. Choudhury, J. Chutia, A. R. Pal, N. N. Dass, D. Devi and D. S. Patil, Appl. Surf. Sci., 2011, 258, 126 CrossRef CAS PubMed.
- H. Puliyalil, U. Cvelbar, G. Filipič, A. D. Petrič, R. Zaplotnik, N. Recek, M. Mozetič and S. Thomas, RSC Adv., 2015, 5, 37853 RSC.
- K. Fatyeyeva, A. Dahi, C. Chappey, D. Lanqevin, V. Jean-Marc, F. Poncin-Epaillard and S. Maqais, RSC Adv., 2014, 4, 31036 RSC.
- A. Haji and S. S. Qavamnia, Fibers Polym., 2015, 16, 46 CrossRef CAS.
- C. X. Jia, P. Chen, B. Lin, Q. Wang, C. Lu and Q. Yu, Surf. Coat. Technol., 2010, 204, 3668 CrossRef CAS PubMed.
- C. X. Wang, M. Du, J. C. Lv, Q. Q. Zhou, Y. Ren, G. L. Liu, D. W. Gao and L. M. Jin, Appl. Surf. Sci., 2015, 349, 333 CrossRef CAS PubMed.
- W. B. Liu, S. Zhang, L. F. Hao, W. C. Jiao, F. Yang, X. F. Li and R. G. Wang, Polym. Compos., 2013, 34, 1921 CrossRef CAS PubMed.
- N. Graupner, J. Rossler, G. Ziegmann and J. Mussig, Composites, Part A, 2014, 63, 133 CrossRef CAS PubMed.
- C. X. Wang, H. L. Xu, Y. Liu and Y. P. Qiu, Surf. Coat. Technol., 2008, 202, 2775 CrossRef CAS PubMed.
- M. G. Mccord, Y. J. Hwang, P. J. Hauser, Y. Qiu, J. J. Cuomo, O. E. Hankins and M. A. Bourham, Text. Res. J., 2002, 72, 491 CrossRef CAS PubMed.
- C. X. Wang, Y. Liu, H. L. Xu, Y. Ren and Y. P. Qiu, Appl. Surf. Sci., 2008, 254, 2499 CrossRef CAS PubMed.
- Y. Y. Sun, Q. Liang, H. J. Chi, Y. J. Zhang, Y. Shi, D. N. Fang and F. X. Li, Fibers Polym., 2014, 15, 1 CrossRef CAS.
- P. J. de Lange, P. G. Akker, E. Mäder, S. L. Gao, W. Prasithphol and R. J. Young, Compos. Sci. Technol., 2007, 67, 2027 CrossRef CAS PubMed.
- S. Farris, L. Introzzi, P. Biagioni, T. Holz, A. Schiraldi and L. Piergiovanni, Langmuir, 2011, 27, 7563 CrossRef CAS PubMed.
- G. O. Berim and E. Ruckenstein, Nanoscale, 2015, 7, 3088 RSC.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 










