
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-aqueous template-assisted synthesis of mesoporous nanocrystalline silicon orthophosphate†
Ales
Styskalik
ab,
David
Skoda
ab,
Zdenek
Moravec
a,
Pavla
Roupcova
c,
Craig E.
Barnes
d and
Jiri
Pinkas
 *ab
*ab
aMasaryk University, Department of Chemistry, Kotlarska 2, CZ-61137 Brno, Czech Republic. E-mail: jpinkas@chemi.muni.cz; Fax: +420549492443; Tel: +420549496493
bCEITEC MU, Masaryk University, Kamenice 5, CZ-62500 Brno, Czech Republic
cCEITEC BUT, Brno University of Technology, Technicka 10, 616 00 Brno, Czech Republic
dUniversity of Tennessee, Department of Chemistry, Knoxville, TN 37996-1600, USA
First published on 21st August 2015
Abstract
The first synthesis of mesoporous nanocrystalline silicon orthophosphate Si5P6O25 is presented. The synthetic procedure is based on the non-hydrolytic sol–gel reaction in the presence of Pluronic P123 template and subsequent calcination in air. The condensation of silicon acetate, Si(OAc)4, and tris(trimethylsilyl)phosphate, OP(OSiMe3)3 (TTP), in non-aqueous solvents driven by elimination of trimethylsilyl acetate provides a homogeneous network with a high content of Si–O–P bonds and SiO6 moieties. After burning out the template, mesoporous silicon orthophosphate was obtained with surface areas up to 128 m2 g−1 and pore sizes around 20 nm. The nanocrystalline Si5P6O25 phase forms relatively easily (500 °C, 4 h) in comparison with other synthetic routes. All samples were characterized by SEM, TEM, elemental analysis, TGA, nitrogen adsorption, SAXS, 1H, 13C, 29Si, and 31P solid-state NMR spectroscopy, and powder XRD. These xerogels showed superior catalytic activity and selectivity in methylstyrene dimerization.
Introduction
Silicophosphate compounds are widely used as advanced technology materials. One of their key features is stable Brønsted acidity at elevated temperatures which makes them suitable for applications in fuel cells as proton conducting electrolytes1,2 and in heterogeneous catalysis.3–6The active centers in the solid phosphoric acid catalyst (SPA) are acidic P–OH groups. These arise from various (silico)phosphoric acids (ortho, pyro, poly) which are occluded in the pores or attached at the surface of support phases, such as crystalline silicon phosphates.5 SPA catalysts are considered acidic without any redox effects and propene oligomerization is often used as a test reaction in industrial applications. Many reaction parameters influence catalytic activity and specificity, one of them being the content of water in feedstock. Water slowly hydrolyzes crystalline silicon phosphates (support of the catalyst) and restores P–OH groups on the surface.4 Active phosphate species are continuously washed out in the form of phosphoric acids and catalysts without this type of refreshing (e.g. silica gel impregnated with H3PO4) show short lifetimes.4
The synthesis of silicophosphate compounds and SPA catalysts can be accomplished by high temperature reactions,7–9 chemical vapor deposition,10 or by hydrolytic2,11–13 and non-hydrolytic sol–gel techniques. However, reproducibility can be problematic for the high temperature synthesis from silica and P4O10, H3PO4, or ammonium phosphates as these reactions produce many different crystalline compounds, such as silicon pyrophosphate, SiP2O7 (in eight structural polymorphs) and silicon orthophosphate, Si5O(PO4)6.8,9,14–16 Si–O–P bonds are only formed by the condensation of Si–OH and P–OH on heating these SiO2–H3PO4 heterogeneous mixtures and crystallization to various silicophosphate phases ensues above about 150 °C.17–19 Similar to high temperature syntheses, the hydrolytic sol–gel method is not easily controlled. Silicophosphate xerogels prepared from hydrolyzed TEOS and H3PO4 with a 1P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2Si molar ratio display a signal for isolated orthophosphate in the 31P MAS NMR spectra of heat treated samples even up to 600 °C where it coexists with crystalline silicophosphate phases containing octahedral SiO6 units.20
:2Si molar ratio display a signal for isolated orthophosphate in the 31P MAS NMR spectra of heat treated samples even up to 600 °C where it coexists with crystalline silicophosphate phases containing octahedral SiO6 units.20
Previously, we have shown than non-hydrolytic sol–gel reactions based on ester elimination allow the preparation of amorphous, microporous silicophosphate xerogels with high homogeneity on atomic level.21 The crystallization of silicon orthophosphate from these xerogels was observed at low temperatures and in short calcination times by HT-XRD measurements (150 °C, 1.5 h). However the resulting material was nonporous.21 Since a goal of our research is to synthesize new heterogeneous catalysts, we need to prepare thermally stable silicophosphates with high surface areas and preferably large pores (mesopores) that make acid sites accessible to broad range of substrates. Recently we succeeded in obtaining mesoporous organosilicophosphates by introducing bridging acetoxysilanes.22 These hybrid xerogels exhibit surface area up to 700 m2 g−1 and maximum pore size of 9.0 nm.
Block copolymers act as templates in aprotic organic solvents and provide short range order in metal oxides with wormhole pore structure.23–26 In contrast, examples of ordered phosphate preparation are scarce. Here we report a novel synthesis of mesoporous nanocrystalline silicon orthophosphate by the non-hydrolytic sol–gel condensation in the presence of block copolymer Pluronic P123 and subsequent calcination in air. Silicon tetraacetate, Si(OAc)4, (Ac = CH3CO) and tris(trimethylsilyl)phosphate, OP(OSiMe3)3, (TTP) are used as precursors. The condensation reaction provides a clear gel at ambient temperature in 1–2 hours. The high homogeneity of Si and P mixing on an atomic level is a key property of these xerogels, which makes them easy to reproducibly transform into the crystalline silicon orthophosphate under mild conditions (4 h, 500 °C). Pluronic P123 acts in the synthesis as a porogen and after being burned out provides mesoporosity in calcined nanocrystalline silicon orthophosphate. The surface is covered with strongly acidic P–OH groups. Heterogeneous catalytic activity was tested in methylstyrene dimerization.3–5
Experimental
General synthetic procedures and characterization methods are described in the ESI.†Non-hydrolytic sol–gel reactions
In a typical reaction (Scheme 1), TTP (4.864 g; 15.48 mmol) was added dropwise within few minutes to a stirred solution of Si(OAc)4 (3.431 g; 12.98 mmol) in toluene (50 cm3) at 80 °C under nitrogen. The molar ratio of precursors was 5Si:6P (to be the same as in silicon orthophosphate Si5P6O25). After the addition was complete, the reaction mixture was vigorously stirred for 30 min and then a solution of Pluronic P123 (1.750 g) in toluene (5.25 cm3) was added and the resulting solution was stirred for additional 30 min. The condensation reaction provided a clear colorless gel in 1 h. The gel was kept at 80 °C for one week during which time syneresis occurred. The gel was then dried under vacuum for 2 d and 5.679 g of a white xerogel (SiP–P) was obtained which was subsequently calcined in ambient atmosphere at 500 °C for 4 h (SiP–P-500). The samples after calcination were cooled in dry air to exclude the hydrolysis of Si–O–P bonds. The silicophosphate sample without template (SiP) was synthesized for the sake of comparison in a similar manner without Pluronic P123.
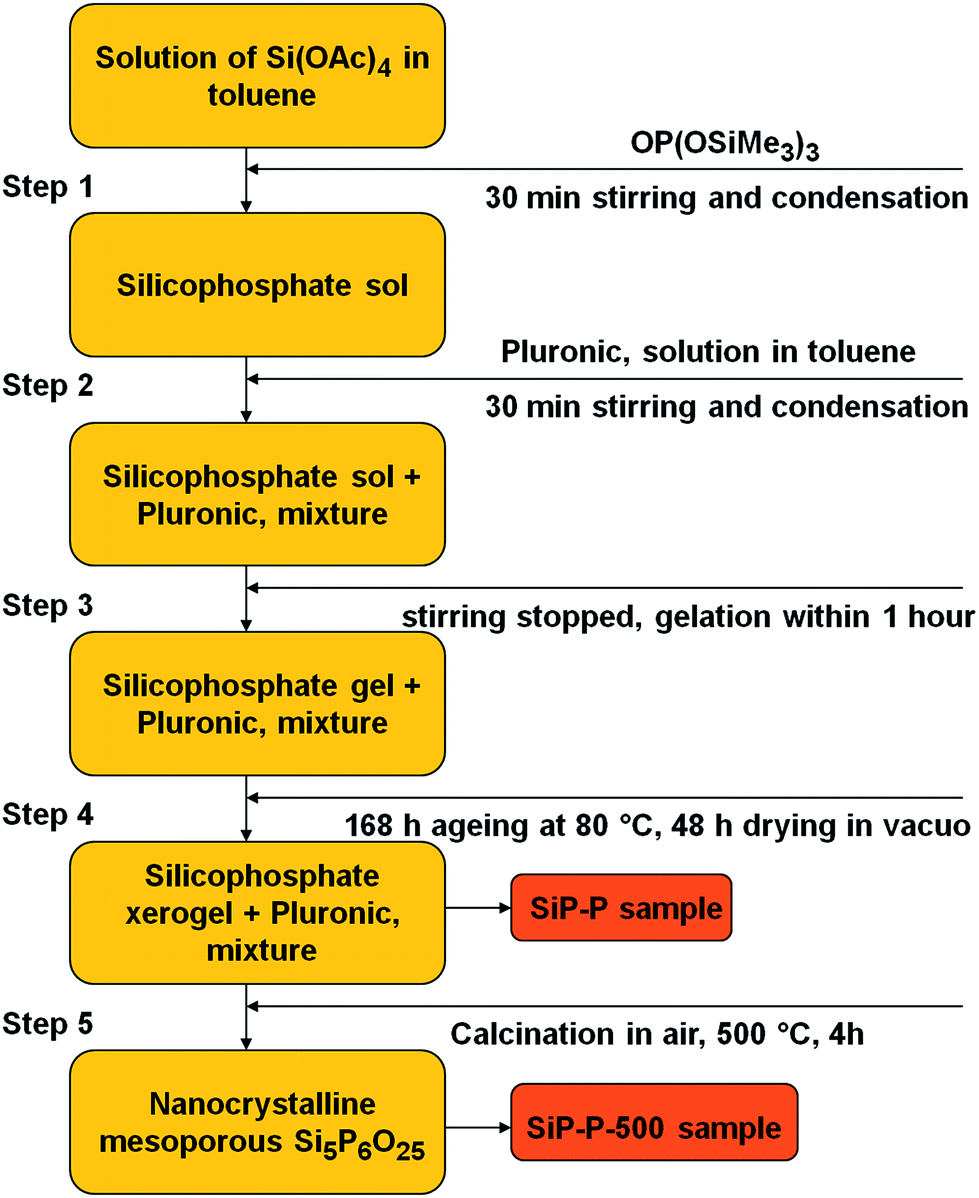 | ||
| Scheme 1 Synthetic procedure for the preparation of mesoporous silicophosphates. | ||
The degree of condensation, DC (%) (eqn (1)), estimated from gravimetric analyses (precise weighing of the yield of the product and of the mass of starting precursors), represents the relative difference between the maximum theoretical loss of AcOSiMe3 in comparison to what is experimentally observed. This difference also defines the number of acetoxy groups on silicon and trimethylsilyl ester groups on phosphorus that are left in the matrix. ntotal in eqn (1) means the total molar amount of organic groups in the reaction mixture and nresidual is a molar amount of residual organic groups in the xerogel computed from difference of theoretical and experimental yield. The theoretical DC of 100% would correspond to product formula of Si5P6O24(OAc)2 (eqn (2)). However the condensation reactions were never quantitative, therefore DC was used for calculating the theoretical mass loss, phosphorus and silicon content (w(P)%, w(Si)%).
| DC = (ntotal − nresidual)/ntotal | (1) |
Characterization and analyses
IR (KBr, cm−1) ν: 498 m, 652 w (ν SiC3), 762 w (ρs SiCH3), 853 m (ρas SiCH3), 1021 vw (ρ CH3), 1050 sh (ν Si–O–P), 1115 vs (ν P–O–Si + δ C–O–C Pluronic), 1258 s (δs SiCH3), 1300 sh (ν P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1377 w (δs CH3), 1460 w (δas CH3), 1542 m (νs COO bidentate), 1707 vw (νas COO), 1766 m (νas COO monodentate), 2870 m (νs CH2) 2907 vw (νs CH3), 2934 m (νas CH2), 2969 w (νas CH3).
O), 1377 w (δs CH3), 1460 w (δas CH3), 1542 m (νs COO bidentate), 1707 vw (νas COO), 1766 m (νas COO monodentate), 2870 m (νs CH2) 2907 vw (νs CH3), 2934 m (νas CH2), 2969 w (νas CH3).
13C CPMAS NMR (ppm) δ: −0.5 (POSiCH3), 16.0 (CH3COO bident + O–CH(CH3)CH2– Pluronic), 21.2 (CH3COO unident), 69.8 (O–CH2CH2– Pluronic), 72.6 (O–CH(CH3)CH2– Pluronic), 74.4 (O–CH(CH3)CH2– Pluronic), 165.8 (CH3COO unident), 190.1 (CH3COO bident).
29Si CPMAS NMR (ppm) δ: 25.0 (POSiMe3), −108 (SiO4), −196 (SiO6), −216 (SiO6).
31P MAS NMR (ppm) δ: −41 (P(OSi)4), −31 (OP(OSi)3).
IR (KBr, cm−1) ν: 409 w, 476 m, 498 w, 553 m, 643 w, 723 w, 807 w, 1030 vs, 1110 vs, 1162 vs, 1611 w (δ OH), 1735 w, 3449 w (ν OH).
29Si CPMAS NMR (ppm) δ: −104 (SiO4), −119 (SiO4), −213 (SiO6), −217 (SiO6).
31P MAS NMR (ppm) δ: −44.7 (P(OSi)4), −20.7 (P(OSi)2(OH)2). ICP-OES (%): calcd Si 19.3, P 25.6. Exp. Si 19.2, P 24.3.
The organic product of the condensation reactions – trimethylsilylacetate, AcOSiMe3 – was identified by a combined GC-MS technique with signals at m/z 45/61/75/117.
α-Methylstyrene dimerization
A suspension of SiP–P-500 (5 wt%) in dry α-methylstyrene (2.0 g) was prepared and the mixture heated to 90 °C for 6 h.6 The silicophosphate catalyst was then filtered off and, after dilution with toluene (1:20 vol), the product mixture was analyzed by GC-MS. The results were estimated on the basis of relative ratios of integrals of peaks in chromatograms, assuming that the different α-methylstyrene dimers (Scheme S1†) will provide similar ion currents. The catalyst reusability was tested after separating it from the reaction mixture by centrifugation, washing three times with toluene (10 cm3), and drying in flowing nitrogen at room temperature. The dried catalyst was used again for the next α-methylstyrene dimerization.
Results and discussion
Silicophosphate xerogels (SiP–P samples) were synthesized by the ester elimination route from silicon acetate and TTP (eqn (2)) in toluene as reported previously.21 The synthetic protocol was modified only in two parameters: (i) the Si:P ratio was set to 5:6 to correspond to silicon orthophosphate Si5P6O25 and (ii) a templating agent Pluronic P123 was added into the reaction mixture after 30 min of condensation (Scheme 1). Gelation was achieved in the presence of Pluronic P123 and the expected condensation byproduct AcOSiMe3 (eqn (2)) was observed by GC-MS analysis of the reaction volatiles.
A very small amount of acetic acid was observed in volatiles. It was probably formed by the reaction of terminal hydroxyl groups of templating agent with silicon tetraacetate (initial OH:OAc ratio in starting materials was approximately 1:100).
Ester elimination reactions generally proceed to condensation degrees (DC) of 78.7–84.7% (eqn (3) represents DC = 84.7%) which is comparable to silicophosphate xerogels without Pluronic P123 (SiP samples).21 Incomplete condensation resulted in the presence of unreacted organic groups (eqn (3), and discussion of IR and MAS NMR spectra below) which, upon calcination led to thermogravimetric mass losses of 57.6–60.5% at 1000 °C (Fig. 1). These mass losses are in good agreement with theoretical values computed from DC (see Experimental) and higher than observed in template-free SiP samples (29.5–40.4%), the difference being the combustion of the Pluronic templating agent (Fig. 1).
| 5Si(OAc)4 + 6OP(OSiMe3)3 → Si5P6O24(OAc)2 + 18AcOSiMe3 | (2) |
| 5Si(OAc)4 + 6OP(OSiMe3)3 → Si5P6O21.3(OAc)2+2.7(OSiMe3)2.7 + 15.3AcOSiMe3 | (3) |
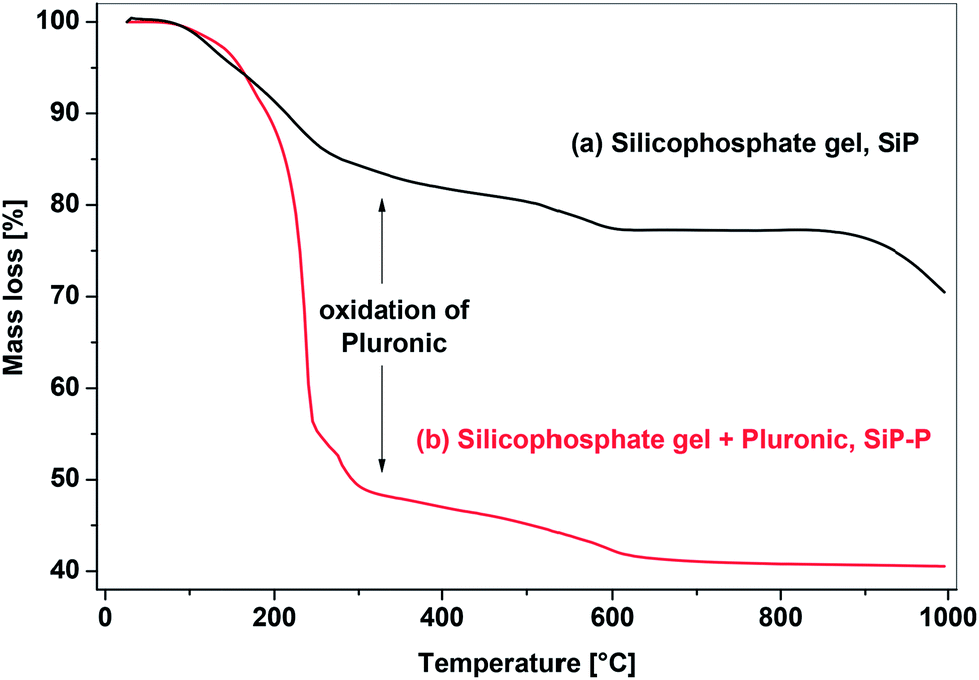 | ||
| Fig. 1 TG analyses of SiP and SiP–P samples, heating rate 5 °C min−1, to 1000 °C. | ||
As already presented above, the Pluronic template did not hinder the condensation reaction and the same conclusion was made for the structure of the xerogels after the examination of IR and MAS NMR spectra. The infrared spectra of SiP and SiP–P samples (Fig. 2) exhibit strong bands at 1100 cm−1, which is typical for homogeneous silicophosphate glasses27,28 as well as for parent homogeneous silicophosphate xerogels (SiP)21 and serves as a benchmark for the Si–O–P bonds.12,13,17,29,30 The presence of residual trimethylsilyl and acetoxy groups was gleaned from strong bands located at 762, 853, and 1258 cm−1 (Me3SiO) and 1021, 1542, 1707, and 1766 cm−1 (AcO) in both samples as well. The only significant differences between the IR spectra of SiP–P and SiP samples (Fig. 2) are absorption bands located at 1115, 1348, 2870, and 2934 cm−1 which we assign to ν(C–O–C), ν(C–C), νs(CH2) and νas(CH2), respectively, from the templating agent Pluronic P123.31 To conclude, the IR spectra suggest that SiP–P sample is a mixture of silicophosphate xerogel with Pluronic. There are no hints of changes in the structure, destruction of the Si–O–P skeleton or lowering the homogeneity.
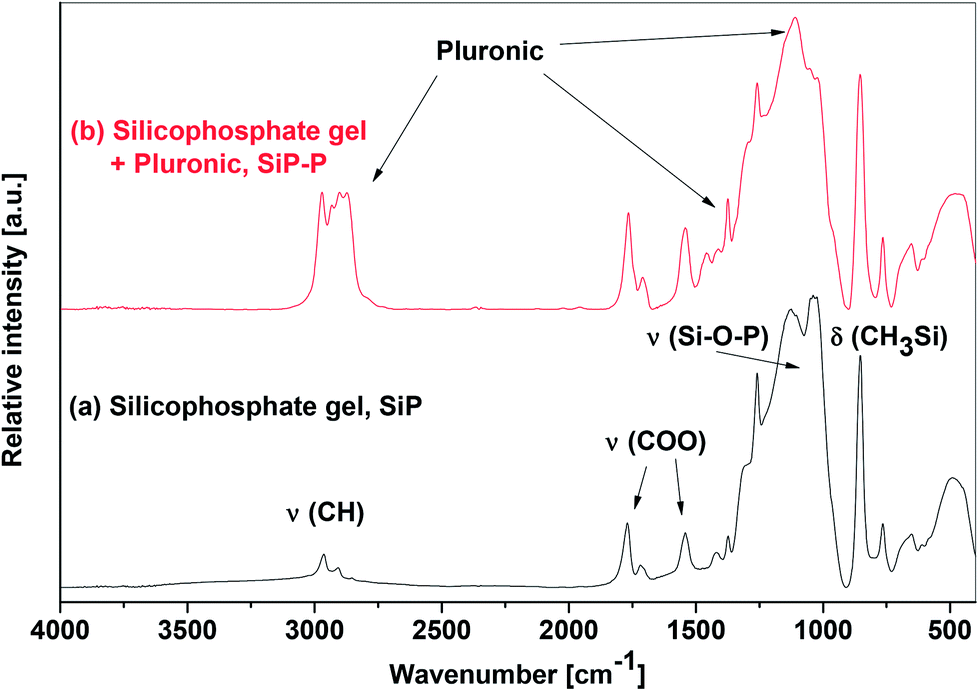 | ||
| Fig. 2 IR spectra of SiP and SiP–P xerogels in KBr pellets. | ||
The conclusions drawn from the IR spectra were confirmed by MAS NMR spectroscopy. The 29Si CPMAS spectrum of SiP–P sample (Fig. S1†) consisted of four resonances at 25.8 ppm (unreacted Me3SiO), −108 ppm (SiO4), −196 ppm (SiO6, additional coordination of carboxyls of residual acetoxy groups), and −216 ppm (SiO6, additional coordination of phosphoryl groups) which exactly matched the ones observed in our parent silicophosphate xerogels (SiP, Fig. S1†).2131P MAS NMR spectrum (Fig. 3, bottom) displayed only one broad resonance with maximum at −40 ppm typical for P(OSi)4 moieties21,22 with a shoulder at −31 ppm assigned to OP(OSi)3 groups.32–34 New signals (in contrast to SiP samples) were observed only in 13C CPMAS NMR spectra (Fig. S2†) – in addition to the signals of Me3SiOP groups (−0.5 ppm), unidentate AcO groups (21.2 and 165.8 ppm) and bidentate AcO groups (16.0 and 190.1 ppm), the signals of polyethylene oxide–polypropylene oxide copolymer were present at 16.0 ppm (methyl group, O–CH(CH3)CH2), 69.8 ppm (O–CH2CH2), 72.6 ppm (O–CH(CH3)CH2) and 74.4 ppm (O–CH(CH3)CH2).35,36 The MAS NMR spectra revealed, that the important structural units, such as SiO6(carboxyl), SiO6(phosphoryl), P(OSi)4, Me3SiOP, uni- and bidentate acetates of the parent silicophosphate were retained within the templated SiP–P samples. No influence of Pluronic on either condensation or structure was observed and thus we started to follow the thermal transformations of SiP–P xerogels.
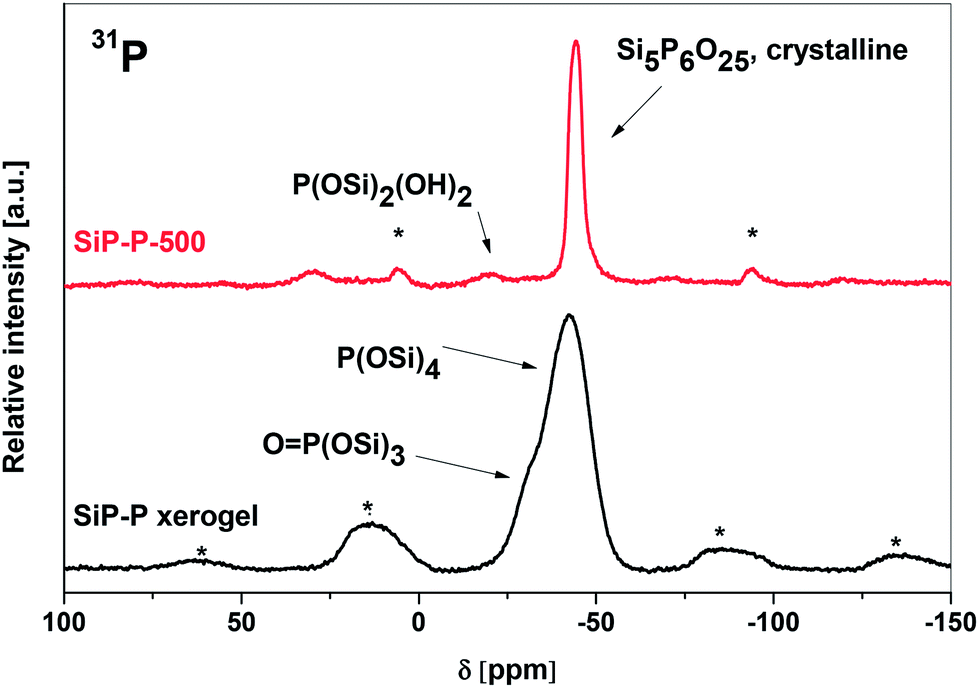 | ||
| Fig. 3 31P MAS NMR spectra of SiP–P xerogel (bottom) and SiP–P-500 sample (upper). Rotation rate 10 kHz, * marks spinning sidebands. | ||
As reported previously, silicon orthophosphate Si5P6O25 starts to crystallize from the SiP xerogels already at 150 °C,21 however there are several other processes that occur during the calcination of the SiP–P samples – oxidation of Pluronic, oxidation of residual trimethylsilyl groups and hydrolysis of resulting silicon phosphate in moist air. We used the temperatures from 500 to 700 °C for the calcination of SiP–P samples as at lower temperatures black color was formed. We tested the influence of moisture in the atmosphere during the calcination and found that trimethylsilyl groups are more easily eliminated as trimethylsilanol or hexamethyldisiloxane in ambient atmosphere, while the calcination in dry synthetic air leads mostly to their oxidation to SiO2, which is then present as an admixture in the samples (as revealed by 29Si CPMAS and ICP-OES analysis of Si and P). Finally, the resulting silicon orthophosphate was found to partially hydrolyze during cooling in ambient atmosphere (31P MAS, XRD). Therefore we calcined the samples to 500 °C for 4 h (heating ramp 5 °C min−1) in ambient atmosphere and then we let the samples cool down in dry atmosphere (SiP–P-500). Following this procedure we reproducibly obtained white crystalline samples, with only a small amount of SiO2 (5–8%), and silicon orthophosphate was not hydrolyzed (Scheme 1, step 5).
The XRD analysis of the calcined samples (SiP–P-500) unambiguously proved that the resulting white powder can be described as silicon orthophosphate, Si5P6O25 (PDF 40-457). No evidence of any other crystalline phases was observed and the content of amorphous phase was estimated to be 18 wt%. The composition of amorphous phase is discussed below. The Bragg peaks for Si5P6O25 are broad (Fig. 4) suggesting these materials are made up of small crystallites, which had an average size of 16 nm based on the Debye–Scherrer equation. It is notable that the formation of crystalline phase is relatively fast in comparison with other procedures, where the crystallization of Si5P6O25 has only been observed after melting silica gel and phosphoric acid for several days.7,12,13,19 This effect is caused by homogeneous Si/P mixing at the molecular level, which is the major advantage of our non-hydrolytic sol–gel technique (Scheme 1, eqn (2)).
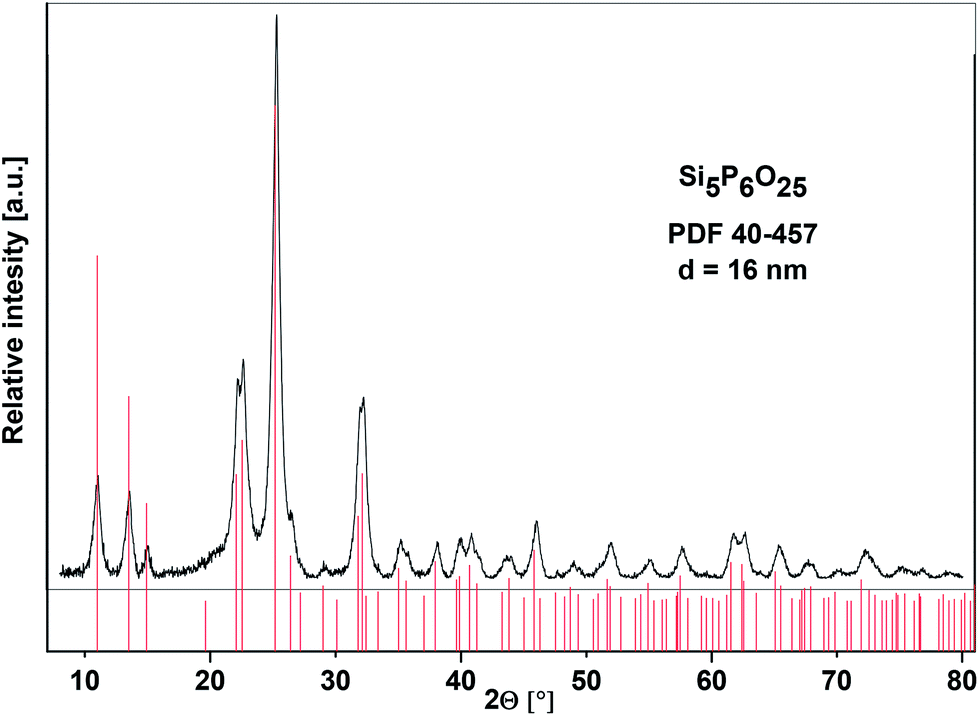 | ||
| Fig. 4 XRD diffractogram of SiP–P-500 sample and corresponding PDF card of silicon orthophosphate. | ||
The crystal structure of silicon orthophosphate consists of three distinct Si sites – two octahedral and one tetrahedral14,16 consistent with what is observed in the 29Si CPMAS NMR spectrum (Fig. S1,† resonances at −119, −213, and −217 ppm). Chemical shifts are in the good agreement with the published data.33,34 These results together with the XRD analysis lead to the conclusion that SiP–P-500 consists predominately of Si5P6O25. There is an additional signal at −104 ppm observed in the 29Si CPMAS NMR spectrum (Fig. S1†). This could be ascribed to amorphous silica33 which is formed by partial oxidation of the Me3SiO groups during the calcination of the SiP–P samples. As already discussed, the silica content is lower if the calcination is performed in ambient (moist) atmosphere instead of dry air.
There is only one type of phosphate group present in the structure of silicon orthophosphate represented by a resonance at −44.7 ppm in the 31P MAS NMR spectrum (Fig. 3, upper), again in a very good agreement with published data.33,34 This major signal is accompanied by a peak of very low intensity at −20.7 ppm, which could be assigned to P(OSi)2(OH)2 or OP(OH)(OSi)2 according to the recent literature37,38 and arises probably from the surface phosphate groups which are terminated with hydroxyl groups. The formation of Brønsted acidic P–OH groups on the surface was independently confirmed by a peak at 8.5 ppm in the 1H MAS NMR spectrum of SiP–P-500.
The IR spectra of SiP–P-500 were recorded in order to establish the nature of structural units present in the calcined samples. A typical spectrum is shown in Fig. 5a and displays absorption bands of valence vibrations (900–1350 cm−1) and deformation vibrations (400–850 cm−1) of Si–O–P and Si–O–Si bonds.9,39 The recorded spectra agree well with the data reported by Malshikov for crystalline silicon orthophosphate, Si5P6O25.9 There is also a broad band at 3400 cm−1 of OH valence vibrations which corroborates the presence of surface hydroxyl groups. The IR spectra of pyridine-treated sample (Fig. 5b) displayed in additional absorption bands of pyridinium cation40,41 at 1490, 1542, and 1638 cm−1 and confirmed Brønsted acidity of silicon orthophosphates.
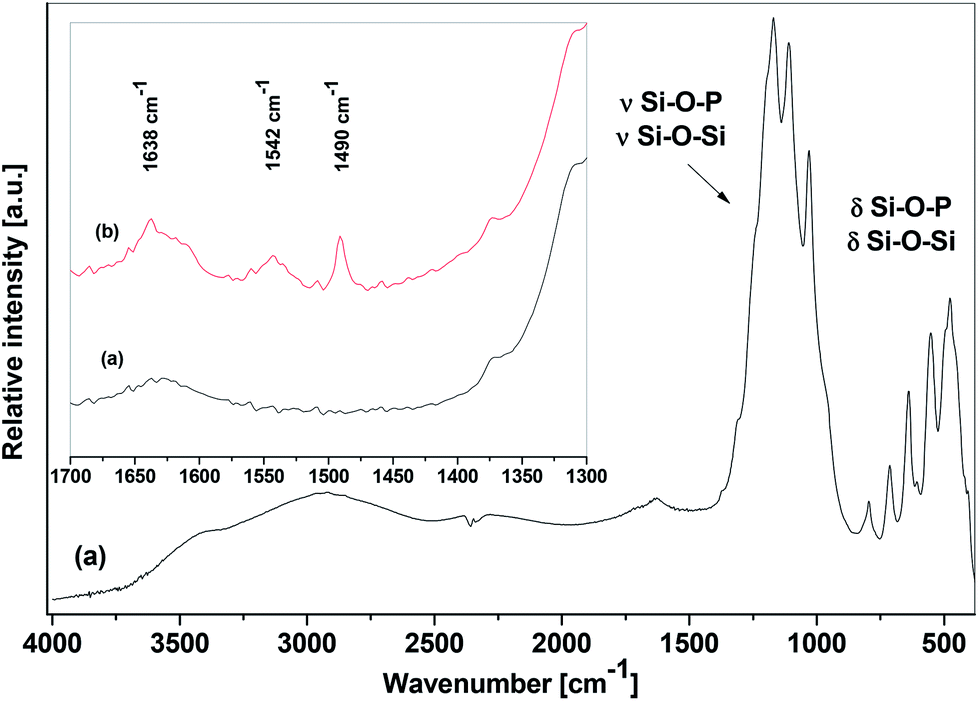 | ||
| Fig. 5 IR spectra of SiP–P-500 sample stored in dry atmosphere (a) and exposed to pyridine at its autogeneous pressure (b). | ||
Useful information about the amount of silicon dioxide admixture was gained by the ICP-OES analysis of Si and P contents in mineralized samples. The nominal Si:P ratio 5:6 changed to the benefit of silicon. It was not caused by the loss of phosphorus at higher temperatures,21 which is frequently observed at the silicophosphate products of hydrolytic sol–gel syntheses due to the low cross-linking between Si and P,11–13,19 but it was caused by the partial oxidation of trimethylsilyl groups to silica. The Si:P ratio in the SiP–P-500 samples was in the range from 5.2:6 to 5.5:6. In other words, silica formed 5–8 wt% of resulting samples thus being only a minor phase. This value is in broad agreement with quantity of amorphous phase (18 wt%) determined by XRD. The amorphous phase may consist not only of silicon dioxide but also of some small part of non-crystalline silicophosphate.
The degree of ordering was followed by the SAXS measurement. The uncalcined samples (SiP–P) showed one scattering maximum indicating a short range order similar to materials prepared by Kriesel24 in toluene (Fig. S3†). This peak was lost during the calcination to 500 °C (SiP–P-500), probably due to crystallization of the silicon orthophosphate phase (Fig. S3†). Therefore we focused on alternative methods of low temperature removal of Pluronic P123 template, such as ozonolysis, BBr3 cleavage, and oxidation by dry oxygen at 60 °C combined with extraction into CH2Cl2. With these procedures we were able to remove at most 15.8% of the template according to TGA, while the ordered mesostructure was maintained.
The nitrogen adsorption–desorption isotherm of SiP–P-500 with its DFT pore size distribution is shown in Fig. 6. According to IUPAC nomenclature the obtained isotherm is type IV, characteristic of mesoporous materials. Since the steep hysteresis loop is observed in the p/p0 region from 0.8 to 1.0, it could be suggested that the samples contain high degree of textural porosity.24,42 This is in agreement with SAXS measurements of SiP–P-500 sample. The surface areas determined by the BET method ranged from 92 to 128 m2 g−1 for samples from different batches, total pore volumes (at p/p0 = 0.976) reached from 0.503 to 0.595 cm3 g−1 and average pore diameters were in the range from 18 to 22 nm. The DFT pore size distribution was relatively narrow with pore diameters ranging from 10 to 45 nm. Materials with textural porosity usually show wider pore size ranges so the SiP–P-500 sample probably contains certain fraction of structure confined porosity.24 According to the t-plot analyses there were no micropores present in the samples.
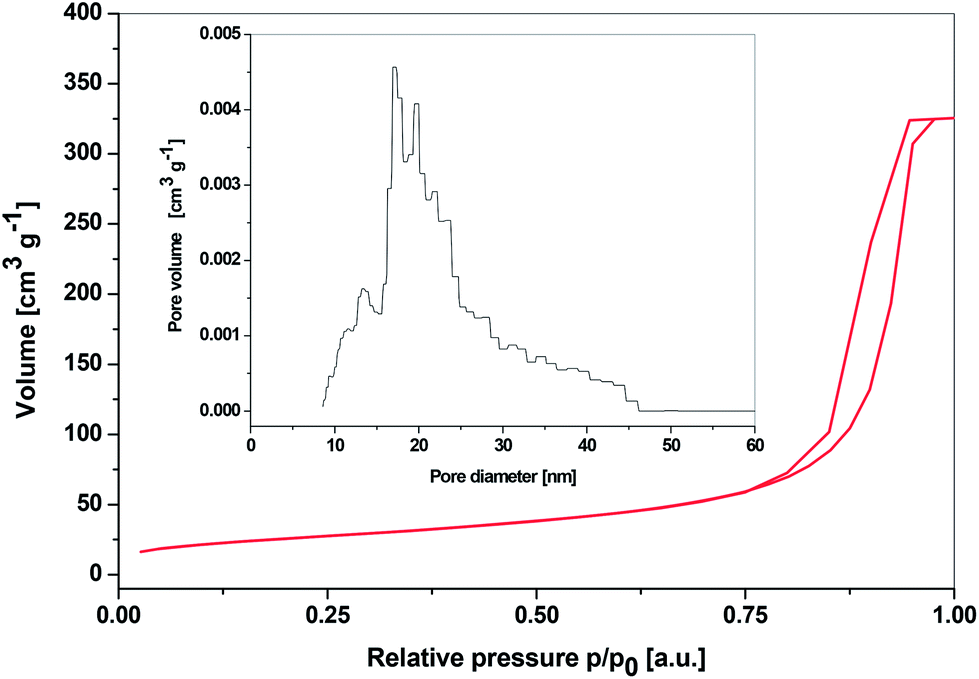 | ||
| Fig. 6 N2 adsorption–desorption isotherms of SiP–P-500 sample and its DFT pore size distribution (inset, DFT Kernel used: N2 at 77 K on silica, cylindrical pore, NSDFT equilibrium model). | ||
These results signal a significant improvement of our materials in the context of heterogeneous catalysis applications. While the parent SiP xerogels were microporous and their heating resulted in nonporous silicon orthophosphate, these new SiP–P-500 samples are thermally stable, possess reasonable high specific surface areas and are mesoporous with relatively narrow pore size distributions. To our knowledge, this is the first example of the synthesis of crystalline and mesoporous silicon orthophosphate.
The SEM and TEM images of calcined silicophosphates prepared with the Pluronic template are shown in Fig. 7. The SEM image shows the vermicular nature of the SiP–P-500 sample where the sponge-like porous structure can be seen and thus corroborates the results of N2 adsorption–desorption experiments. In contrast the SEM image of calcined SiP sample (Fig. S4†) shows agglomerated particles. The agglomeration is probably reason for the low surface area of the sample (<10 m2 g−1). The TEM image shows the primary particles of SiP–P-500 sample. Their sizes are consistent with the results of XRD analyses of crystallite size (16 nm).
 | ||
| Fig. 7 SEM (left) and TEM (right) image of SiP–P-500 sample. | ||
Characteristics of SiP–P-500 sample (mesoporosity, Brønsted acidity) suggest that these materials could serve as solid phosphoric acid catalysts. Therefore we performed catalytic tests on α-methylstyrene dimerization, which is catalyzed by acidic protons.6 Various homogeneous and heterogeneous Brønsted acids were tested recently and the best results among heterogeneous catalysts were achieved with phosphoric acid impregnated on silica (conversion 97.5%, selectivity 94.7%, 2–5 wt% of catalyst in neat methylstyrene, 6 h, 90 °C).6 We used the same conditions for this reaction and the SiP–P-500 sample with the surface P–OH groups performed the catalytic reaction with up to 99% conversion of α-methylstyrene and 99% selectivity toward cyclic dimer I (Scheme S1,† GC-MS). The reused catalyst maintained very high conversion of 99% in the second catalytic run. Meantime the selectivity only slightly decreased to 98%. Heterogeneous nature of catalytic action is supported by the absence of phosphorus in the reaction solution (ICP-OES). We compared the catalytic action of porous sample SiP–P-500 with nonporous crystalline sample prepared without a template (SiP-500) and found out that the latter reached almost complete conversion of methylstyrene after 6 h at 90 °C as well. Therefore we decreased the reaction time and temperature to 5 min and 50 °C. While conversion remained high for SiP–P-500 (98%), it dropped to 89% for SiP-500. Selectivity changed for both catalysts to 47 and 45% of terminal dimer (II).
Our catalysts possess a large amount of phosphorus species dispersed homogeneously on the surface. Therefore we believe, that these mesoporous nanocrystalline silicon phosphates would show an increased stability in comparison with phosphoric acid impregnated on silica.
Conclusions
All the data in hand showed that our synthetic procedure relying on non-hydrolytic polycondensation between silicon tetraacetate, Si(OAc)4, and tris(trimethylsilyl)phosphate, OP(OSiMe3)3, in the presence of templating agent Pluronic P123 and subsequent short calcination resulted in the formation of nanocrystalline, mesoporous silicon orthophosphate, Si5P6O25, with surface areas up to 128 m2 g−1 and pore sizes around 20 nm. The porosity is mainly of textural type. The short calcination time needed for the crystallization to silicon orthophosphate Si5P6O25 was achieved through the atomic homogeneity of silicophosphate xerogels with high content of Si–O–P bonds resulting from the non-hydrolytic condensation reaction. The nanocrystalline and mesoporous silicon orthophosphate was covered with surface P–OH groups providing Brønsted acidity. These xerogels showed superior catalytic behavior in methylstyrene dimerization and are promising materials for potential heterogeneous catalysis applications.Acknowledgements
Authors thank the project CEITEC – Central European Institute of Technology CZ.1.05/1.1.00/02.0068, Structural Analysis Laboratory of CEITEC, and KONTAKT II LH11028 for the financial assistance. J. P. thanks to the Fulbright Foundation for a scholarship. A. S. thanks to the Brno City Municipality for Brno PhD Talent Scholarship. Authors thank Dr K. Novotny and L Simonikova for ICP-OES analyses, Dr M. Klementova for TEM analyses and T. Samoril for SEM images.References
- M. Mika, M. Paidar, B. Klapste, M. Masinova, K. Bouzek and J. Vondrak, J. Phys. Chem. Solids, 2007, 68, 775–779 CrossRef CAS.
- A. Matsuda, T. Kanzaki, K. Tadanaga, M. Tatsumisago and T. Minami, Solid State Ionics, 2002, 154–155, 687–692 CrossRef CAS.
- C. M. Fougret and W. F. Hölderich, Appl. Catal., A, 2001, 207, 295–301 CrossRef CAS.
- F. Cavani, G. Girotti and G. Terzoni, Appl. Catal., A, 1993, 97, 177–196 CrossRef CAS.
- T. R. Krawietz, P. Lin, K. E. Lotterhos, P. D. Torres, D. H. Barich, A. Clearfield and J. F. Haw, J. Am. Chem. Soc., 1998, 120, 8502–8511 CrossRef CAS.
- G. N. Kirichenko, N. G. Grigoreva, V. I. Glazunova and U. M. Dzhemilev, Petroleum Chemistry, 2009, 49, 306–310 CrossRef.
- F. Liebau, G. Bissert and N. Köppen, Z. Anorg. Allg. Chem., 1968, 359, 113–134 CrossRef CAS.
- H. Makart, Helv. Chim. Acta, 1967, 50, 399–405 CrossRef CAS.
- A. E. Malshikov, Russ. J. Inorg. Chem., 1987, 32, 1383–1385 Search PubMed.
- W. A. Gambling, D. N. Payne, C. R. Hammond and S. R. Norman, Proc. Inst. Electr. Eng., 1976, 123, 570 CrossRef CAS.
- F. Tian, L. Pan, X. Wu and F. Wu, J. Non-Cryst. Solids, 1988, 104, 129–134 CrossRef CAS.
- Y. S. Kim and R. E. Tressler, J. Mater. Sci., 1994, 29, 2531–2535 CrossRef CAS.
- A. Aronne, M. Turco, G. Bagnasco, P. Pernice, M. Di Serio, N. J. Clayden, E. Marenna and E. Fanelli, Chem. Mater., 2005, 17, 2081–2090 CrossRef CAS.
- D. M. Poojary, R. B. Borade and A. Clearfield, Inorg. Chim. Acta, 1993, 208, 23–29 CrossRef CAS.
- D. M. Poojary, R. B. Borade, F. L. Campbell III and A. Clearfield, J. Solid State Chem., 1994, 112, 106–112 CrossRef CAS.
- H. Mayer, Monatsh. Chem., 1974, 105, 46–54 CrossRef CAS.
- P. Massiot, M. A. Centeno, I. Carrizosa and J. A. Odriozola, J. Non-Cryst. Solids, 2001, 292, 158–166 CrossRef CAS.
- C. Fernández-Lorenzo, L. Esquivias, P. Barboux, J. Maquet and F. Taulelle, J. Non-Cryst. Solids, 1994, 176, 189–199 CrossRef.
- S.-P. Szu, L. C. Klein and M. Greenblatt, J. Non-Cryst. Solids, 1992, 143, 21–30 CrossRef CAS.
- A. Matsuda, T. Kanzaki, Y. Kotani, M. Tatsumisago and T. Minami, Solid State Ionics, 2001, 139, 113–119 CrossRef CAS.
- A. Styskalik, D. Skoda, Z. Moravec, J. G. Abbott, C. E. Barnes and J. Pinkas, Microporous Mesoporous Mater., 2014, 197, 204–212 CrossRef CAS.
- A. Styskalik, D. Skoda, Z. Moravec, M. Babiak, C. E. Barnes and J. Pinkas, J. Mater. Chem. A, 2015, 3, 7477–7487 CAS.
- P. T. Tanev and T. J. Pinnavaia, Chem. Mater., 1996, 8, 2068–2079 CrossRef CAS.
- J. W. Kriesel, M. S. Sander and T. D. Tilley, Chem. Mater., 2001, 13, 3554–3563 CrossRef CAS.
- P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Chem. Mater., 1999, 11, 2813–2826 CrossRef CAS.
- D. Skoda, A. Styskalik, Z. Moravec, P. Bezdicka and J. Pinkas, J. Mater. Sci., 2015, 50, 3371–3382 CAS.
- J. Wong, J. Non-Cryst. Solids, 1976, 20, 83–100 CrossRef CAS.
- N. Shibata, M. Horigudhi and T. Edahiro, J. Non-Cryst. Solids, 1981, 45, 115–126 CrossRef CAS.
- H. Niida, M. Takahashi, T. Uchino and T. Yoko, J. Mater. Res., 2003, 18, 1–3 CrossRef CAS.
- Z. Cao, B. I. Lee, W. D. Samuels, L.-Q. Wang and G. J. Exarhos, J. Mater. Res., 1998, 13, 1553–1559 CrossRef CAS.
- F. S. Freitas, J. N. de Freitas, B. I. Ito, M.-A. D. Paoli and A. F. Nogueira, ACS Appl. Mater. Interfaces, 2009, 1, 2870–2877 CAS.
- S. Jähnigen, E. Brendler, U. Böhme and E. Kroke, Chem. Commun., 2012, 48, 7675 RSC.
- C. Coelho, T. Azais, L. Bonhomme-Coury, J. Maquet, D. Massiot and C. Bonhomme, J. Magn. Reson., 2006, 179, 114–119 CrossRef CAS.
- C. Lejeune, C. Coelho, L. Bonhomme-Coury, T. Azaïs, J. Maquet and C. Bonhomme, Solid State Nucl. Magn. Reson., 2005, 27, 242–246 CrossRef CAS.
- H. M. Kao, C. L. Chen and S. W. Chiao, J. Chin. Chem. Soc., 2005, 52, 693–699 CrossRef CAS.
- S. Choe, M. J. Kim, H. C. Kim, T. Lim, K. J. Park, K. H. Kim, S. H. Ahn, A. Lee, S.-K. Kim and J. J. Kim, J. Electroanal. Chem., 2014, 714–715, 85–91 CrossRef CAS.
- S. Jähnigen, E. Brendler, U. Böhme, G. Heide and E. Kroke, New J. Chem., 2014, 38, 744 RSC.
- C. Coelho, T. Azais, C. Bonhomme, L. Bonhomme-Coury, C. Boissière, G. Laurent and D. Massiot, C. R. Chim., 2008, 11, 387–397 CrossRef CAS.
- B. Lelong, Ann. Chim., 1964, 9, 229–260 CAS.
- M. Lefrancois and G. Malbois, J. Catal., 1971, 20, 350–358 CrossRef CAS.
- R. B. Borade and A. Clearfield, Appl. Catal., A, 1992, 80, 59–77 CrossRef CAS.
- K. S. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra10982e |
| This journal is © The Royal Society of Chemistry 2015 |