
Co3O4/C/graphene nanocomposites as novel anode materials for high capacity lithium ion batteries†
Lei Liua,
Dong Fangab,
Ming Jiangab,
Jianping Chena,
Tao Wangac,
Qing Wangd,
Lijie Dongae and
Chuanxi Xiong*ab
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, School of Materials Science and Engineering, Wuhan University of Technology, Luoshi Road 122, 430070, Wuhan, China. E-mail: leiliu@whut.edu.cn; cxiong@whut.edu.cn
bSchool of Materials Science and Engineering, Wuhan Textile University, Yangguang Road 1, 430200, Wuhan, China
cDepartment of Physics and Astronomy, University of Sheffield, Sheffield, S3 7RH, UK
dDepartment of Materials Science and Engineering, Pennsylvania State University, University Park, Pennsylvania 16802, USA
eDepartment of Materials Science and Engineering, Cornell University, Ithaca, New York 14850, USA
First published on 24th August 2015
Abstract
A nanocomposite, consisting of graphene intercalated with Co3O4/C hybrids, was prepared through thermal decomposition and carbonization of the precursor Co(NO3)2·6H2O in solvent-free graphene fluids. Co3O4 forms the core of the hybrids and contains a “soft” interface with the C shell. The Co3O4/C/graphene nanocomposite was explored as the anode material in lithium ion batteries, and exhibited an excellent specific capacity of 1050 mA h g−1, a value that is higher than the theoretical value of Co3O4 (890 mA h g−1) at the current density of 100 mA g−1 after 40 cycles. The Co3O4/C/graphene nanocomposite prepared via this route is easy and feasible, and opens up a new pathway to develop electrode materials for high performing LIBs.
Introduction
Lithium-ion batteries (LIBs) present increasing applications in miniaturized and portable electronic devices due to their high energy density, long life time, free memory-effect and environmental friendliness.1 One interest in advancing high performance LIBs is to develop better electrode materials. Spinel cobalt oxide Co3O4 is considered as an alternative anode for LIBs owing to its high theoretical specific capacity (890 mA h g−1), which is typically two or three times higher than that of graphite.2 Nano-sized Co3O4 electrode materials have shown an excellent lithium storage performance.3 However, because of the mechanical fractures and structural pulverization of Co3O4 anode during the device cycling processes, LIBs with a Co3O4 anode generally present poor electronic conduction in cycles. To improve rate performance, carbon coating has been extensively used in anode materials, because the carbon layers can greatly enhance the electronic conductivity of the electrodes.4,5 Graphene, a monolayer of carbon atoms arranged in a honeycomb network enduing with exceptional electrical, mechanical, optical and surface properties, is widely utilized to prepare various hybrid materials.6 In addition, the multilayer structure with high specific surface areas provide not only more active sites to shorten the transporting length of lithium ions, but also enough space to accommodate large volume expansion during the redox process of the electrodes.7 It is an ideal substrate onto which metal oxides can be deposited to improve the cycling performance of LIBs as well as to reduce electric resistance.The preparation of those electrodes for LIBs involves multiple procedures, including mixing the active materials with polymeric binders and conductive carbon additives, then casting the mixture onto a metal foil. The presence of binders, however, can be detrimental, as they can decrease the electrical conductivity, block the diffusion channels of ion transportation, and increase the polarization of the electrodes, all of which significantly reduce the device performance.8 The elimination of binders could lighten the electrode, as well as simplify the preparation process, and effectively improve the electrochemical performance of LIBs.9 However, it still requires an intensive interfacial interaction between the active material and the conductive additives.
In this communication, we synthesized Co3O4/C hybrid nanoparticles that were intercalated in graphene to obtain Co3O4/C/graphene nanocomposite. The nanocomposite was prepared through a thermal decomposition and carbonization of the precursor Co(NO3)2·6H2O in solvent-free graphene fluids (GF). Herein, GF acted as a supporting matrix for guest-active nanocomposite, which is binder free and highly conductive. Moreover, carbon coatings were introduced on the rough surface of Co3O4 nanoparticles through the carbonization of organic long chain grafted on GF, which could protect the inner particles from cracking. The designed nanocomposite exhibited an excellent electro-chemical performance, with large specific capacity, high rate capability and long cycling stability.
Experimental section
Synthesis of Co3O4/C/graphene and fabrication of LIBs
Firstly, graphene oxide nanosheets were synthesized from natural graphite flakes (2.5 g, NP302, Beijing Nanopowder Science & Technology, China) by a modified Hummers method.10 GF was prepared through a two-step procedure involving covalent grafting of graphene oxide nanosheets by 3-(trihydroxysilyl)propane-1-sulfonic acid (SIT) and subsequently ion exchanging with nonylphenolpoly-oxyethylene ether quaternary ammonium salt (NPEQ), which has been described in our previous work.11,12 Then, 0.5 g Co(NO3)2·6H2O were added into GF, followed by an intense stirring treatment for 0.5 h by the combination of magnetic stirring and ultra-sonication. The Co(NO3)2·6H2O/GF mass ration was varied as 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, 1:2, 1:1, 3:2 and 2:1 to optimize the blending ratio. The binder-free mixture was then coated on nickel foam current collector by doctor blading and calcined in a tube furnace maintaining at 220 °C for 1 h and then 700 °C for 5 h under nitrogen atmosphere. The system was then cooled down to room temperature and the target product was collected. The heating rate was set to 10 °C min−1. The electrode was fabricated using the target product as the active material and pure lithium foil as the counter and reference electrode. The electrolyte was composed of 1 M LiPF6 dissolved in ethylene carbonate (EC)/dimethyl carbonate (DMC)/ethyl methyl carbonate (EMC) (1:1:1 by volume), and Celgard 2300 microporous polypropylene was used as the separator. Cell assembly was carried out in an argon-filled glove box with the concentrations of moisture and oxygen below 0.1 ppm.
:3, 1:2, 1:1, 3:2 and 2:1 to optimize the blending ratio. The binder-free mixture was then coated on nickel foam current collector by doctor blading and calcined in a tube furnace maintaining at 220 °C for 1 h and then 700 °C for 5 h under nitrogen atmosphere. The system was then cooled down to room temperature and the target product was collected. The heating rate was set to 10 °C min−1. The electrode was fabricated using the target product as the active material and pure lithium foil as the counter and reference electrode. The electrolyte was composed of 1 M LiPF6 dissolved in ethylene carbonate (EC)/dimethyl carbonate (DMC)/ethyl methyl carbonate (EMC) (1:1:1 by volume), and Celgard 2300 microporous polypropylene was used as the separator. Cell assembly was carried out in an argon-filled glove box with the concentrations of moisture and oxygen below 0.1 ppm.
For comparison, Co3O4/graphene electrode was also prepared by conventional technology.13 The working electrode was fabricated by mixing the active material (Co3O4/graphene nanocomposite):conductivity agent (carbon black, Super-P):binder (polyvinylidene difluoride, PVDF, Aldrich) with a weight ratio of 80:10:10 in N-methylpyrrolidone (NMP) solvent and then pasting onto nickel foam. The electrode was dried at 120 °C in vacuum for 12 h.
Materials characterizations
The crystalline structure was characterized using a Rigaku D/Max-IIIA X-ray generator (XRD, Germany) with Cu-Kα radiation source (λ = 1.5418 Å). X-Ray Photoelectron Spectroscopy (XPS) patterns were recorded by an X-ray photoelectron spectrometer (Thermo Scientific ESCALAB 250Xi, USA), using Mg-Kα radiation as the excitation source. Infrared (IR) transmission spectra were taken on an FTIR spectrometer (Thermo Nicolet Nexus, America) in the range of 4000 to 500 cm−1 using KBr pellets. The morphology of the samples was observed using scanning electron microscope (SEM) (JEOL JSM-6360LV, Japan), before which a thin Au layer (∼3 nm) had been evaporated on the sample surface to form a conductive film for observation. The crystal structure details were observed by transmission electron microscopy (TEM) (Joel JEM-2001F, Japan). TEM samples were deposited on Cu grids coated with carbon support film.Electrochemical measurement for lithium-ion batteries
To evaluate the electrochemical performance, the galvanostatic charge/discharge in the voltage range of 0.01–3.0 V at different current rates (1 mA g−1 = 1/890C, where C corresponds to complete charge or discharge in one hour) and CV at the scan rate of 0.2 mV s−1 were performed using a BTS8 Neware battery cycler and a CHI660E electrochemical workstation, respectively. To analyze the dynamics of basic electrochemical processes, electrochemical impedance spectrum (EIS) tests were conducted with the workstation by using a sine wave of 5 mV amplitude over a frequency range of 100 kHz to 0.01 Hz. The specific capacity was calculated on the basis of the total composite weight.Results and discussion
The schematic of the fabrication process of the Co3O4/C/graphene nanocomposite is shown in Scheme 1. The as-synthesized GF are viscous like liquid even in the absence of any solvent, as well as possess ultra-high specific surface area, owing to their self-unfolding behavior.14 Co(NO3)2·6H2O and GF were mixed and sonicated at room temperature. During the sonication, Co(NO3)2·6H2O are well dispersed by wrapping around flexible long chain, and then readily decomposed to Co3O4 nanoparticles on the self-unfolded graphene sheets through heat treatment. We found that the carbonized degree of Co3O4 nanoparticles gradually improved by increasing the calcination temperature. The Co3O4/C nanoparticles were deposited on the graphene surfaces with relatively high intensity or intercalated into the graphene layers. | ||
| Scheme 1 Schematic illustration for the fabrication of Co3O4/C/graphene nanocomposite. | ||
The morphology of the Co3O4/C/graphene nanocomposite and Co3O4/C hybrids were characterized by SEM and TEM images, respectively. As seen in Fig. 1a, a typical cross-section of Co3O4/C/graphene nanocomposite exhibits well-ordered and layered structure composed of graphene sheets and Co3O4/C hybrid nanoparticles. This multilayer structure serves as a continuous pathway and builts up a conductive network for electron transportation to improve the reaction kinetics of LIBs.15 Fig. 1b highlights a highly homogeneous distribution of Co3O4/C nanoparticles through avoiding agglomeration enabled by the flow ability of GF, giving rise to an enhanced carrier transport and larger specific surface area of the two-dimensional nanostructures. Apparently, these Co3O4/C hybrid nanoparticles acts as spacers and effectively prevented the re-stacking of individual graphene sheet to avoid the loss of their high active surface area.16
 | ||
| Fig. 1 (a) SEM image of the cross-section. (b) SEM image and (c) EDX pattern of Co3O4/C/graphene nanocomposite. (d) TEM images of Co3O4/C/graphene nanocomposite with carbon and cobalt element mapping image in the inset. | ||
To further investigate the internal structure of Co3O4/C/graphene nanocomposite, we performed X-ray diffraction (XRD), energy-dispersive X-ray (EDX), Fourier transform infrared (FTIR) and element mapping. Fig. 2 shows the XRD spectrum of CO3O4/C/graphene nanocomposite. Most of the diffraction patterns in the spectrum could be indexed to the pure phase of Co3O4 (JCPDS Card 42-1467), as indicated in Fig. 2. The small peak at the 2θ of 25.6° is indexed to the (002) crystal plane of the hierarchical graphene layers. The peak at 42.4° originates from the (100) of graphite layer, and appears more intense because this peak overlaps with a diffraction peak from Co3O4.17 Fig. 1c shows the EDX spectrum of Co3O4/C/graphene nanocomposite. The strong Co peaks in the EDX spectrum belong to the Co3O4 nanoparticles, while the strong C peak originates from graphene and carbon coatings. A weak Si peak in the spectrum suggests the existence of Si, which is induced to graphene from the silane coupling agent SIT during preparation. Fig. S1 (ESI†) shows the FTIR spectra of GF, Co(NO3)2·6H2O, GF/Co(NO3)2·6H2O and Co3O4/C/graphene nanocomposite. The peaks at 1104 and 1027 cm−1 correspond to the characteristic peaks of C–O–Si and Si–O–Si respectively.11 These two characteristic peaks still existed after annealing and no new peaks were observed other than the two weak peaks at 570 and 662 cm−1, which originated from the stretching vibrations of Co–O bond and also proved the formation of Co3O4.18
 | ||
| Fig. 2 XRD patterns of Co3O4/C/graphene nanocomposite in the range of 15 to 80°. | ||
The scanning electron microscopy measurements suggests that the size of Co3O4/C nanoparticles is around 50–70 nm. The lattice fringes are visible in the SEM (Fig. S3, ESI†) and TEM (Fig. 1d) observations. The lattice fringe with a lattice spacing of 0.28 nm (see the inset of Fig. S3b, ESI†) corresponds to the (220) plane of the spinel phase Co3O4.19 An element mapping of a Co3O4 nanoparticle was performed and shows that the core is dominated by Co component, while the shell is mainly C element. There isn't a sharp interface to separate the core and shell from the element mapping, suggesting a “soft” interface in the Co3O4/C hybrid nanoparticles. We attribute the “soft” interface as the results of rough surface of the Co3O4 nanoparticles, and subsequently the interdigitation of the carbon and Co3O4 into the core and shell of the hybrid nanoparticles.
X-ray photoelectron spectroscopy (XPS) measurements were performed to investigate the nature of chemical bonds close to the particle surface (<5 nm). The Shirley background was used to subtract the background signals and the peaks were decomposed by fitting using the mixed Gaussian–Lorentzian model. As can be seen from Fig. 3a, the two strong peaks of C 1s and O 1s dominate the spectrum, indicating that carbon and oxygen elements are the main components of the surface of Co3O4/C/graphene nanocomposite. Although there are a lot of Co atoms in the hybrid particle (Fig. 1c), the carbon layer on the surface of Co3O4 nanoparticles is too thick and prevents the examination of Co element using surface sensitive XPS technique. The nature of chemical bonding between elements can be determined from the values of binding energy (BE). The C 1s spectrum of the hybrid is shown in Fig. 3b. The deconvolution of the spectrum suggests four individual peaks (that associate with different chemical bonds) at the positions of 284.7 eV (C–C), 286.5 eV (C–O), 287.9 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and 289.3 eV (C–CO). In Fig. 3c, the broad peak at 531.9 eV is related to the chemical bonding of O–Si that originates from the carbonization of SIT. There is another broad peak at 781.6 eV in Fig. 3d, which might be due to the interfacial Co–O–Si bonds in Co3O4/C/graphene nanocomposite. These Co–O–Si bonds at the interface serve as binders to link Co3O4 nanoparticles with the conductive graphene. We think that Si locates mainly on the surface of Co3O4/C nanoparticles, since there are only a few Si atoms in the Co3O4/C/graphene nanocomposite. This is evidenced by the weak Si 2p peak at 102.1 eV in the XPS spectrum. The Co 2p signal in Fig. 3a shows spin orbit of 2p1/2 and 2p3/2 components that contains the same chemical information. Therefore, only Co 2p3/2 bands were fitted in this study. The oxidation state of cobalt can be deduced from the deconvoluted XPS spectra of Co 2p3/2. The asymmetrical Co 2p3/2 peak of the as-prepared Co3O4/C/graphene nanocomposite could be further decomposed into two components, which can be assigned to Co3+ and Co2+ from low to high binding energy.20 The formation of Co3O4 can be further confirmed by the O 1s peak with a BE of approximately 530 eV, which corresponds to the oxygen species in Co3O4.21
O) and 289.3 eV (C–CO). In Fig. 3c, the broad peak at 531.9 eV is related to the chemical bonding of O–Si that originates from the carbonization of SIT. There is another broad peak at 781.6 eV in Fig. 3d, which might be due to the interfacial Co–O–Si bonds in Co3O4/C/graphene nanocomposite. These Co–O–Si bonds at the interface serve as binders to link Co3O4 nanoparticles with the conductive graphene. We think that Si locates mainly on the surface of Co3O4/C nanoparticles, since there are only a few Si atoms in the Co3O4/C/graphene nanocomposite. This is evidenced by the weak Si 2p peak at 102.1 eV in the XPS spectrum. The Co 2p signal in Fig. 3a shows spin orbit of 2p1/2 and 2p3/2 components that contains the same chemical information. Therefore, only Co 2p3/2 bands were fitted in this study. The oxidation state of cobalt can be deduced from the deconvoluted XPS spectra of Co 2p3/2. The asymmetrical Co 2p3/2 peak of the as-prepared Co3O4/C/graphene nanocomposite could be further decomposed into two components, which can be assigned to Co3+ and Co2+ from low to high binding energy.20 The formation of Co3O4 can be further confirmed by the O 1s peak with a BE of approximately 530 eV, which corresponds to the oxygen species in Co3O4.21
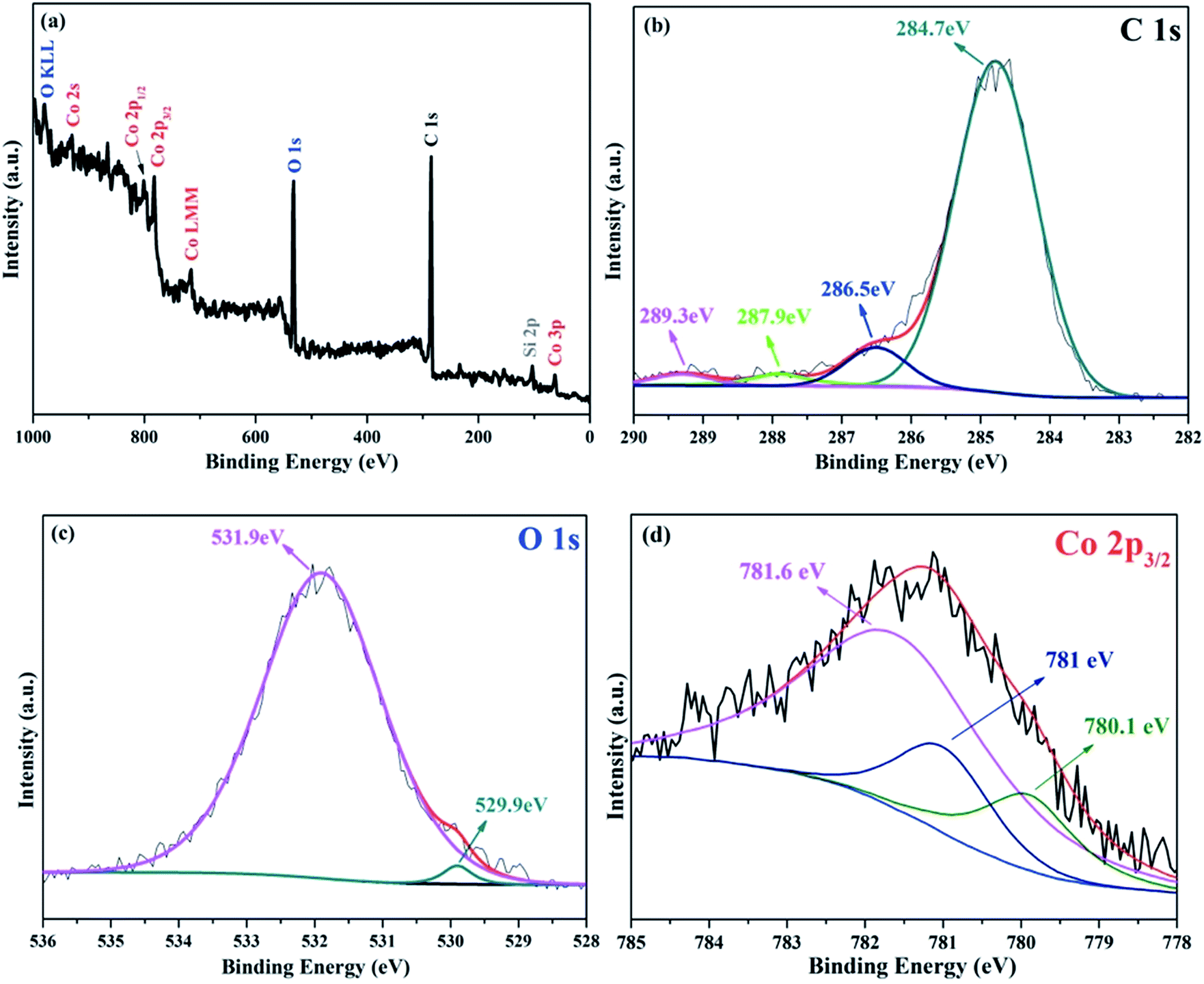 | ||
| Fig. 3 (a) XPS spectrum of the as-prepared Co3O4/C/graphene nanocomposite. Typical C 1s (b), O 1s (c) and Co 2p (d) XPS spectra of Co3O4/C/graphene nanocomposite. | ||
We further investigated the cycle performance of Co3O4/C/graphene nanocomposite at a current density of 100 mA g−1. As shown in Fig. 4a, it demonstrates that a stable and high reversible capacity can be retained at 1050 mA h g−1 after 40 charge–discharges cycles, and is almost 20% higher than the theoretical capacity of spinel Co3O4. The capacity of the lithium storage using Co3O4/C/graphene anode materials that were prepared at different Co(NO3)2·6H2O:GF ratio suggests that the optimal blending ratio is 1:1 (w/w) (Fig. S4†). As a comparison, pure spinel Co3O4 nanoparticles and GF have been prepared and tested under the same condition with Co3O4/C/graphene nanocomposite. The control sample exhibited the average capacities of 340 mA h g−1 and 42 mA h g−1, respectively, as shown in Fig. S5.† This low capacity using Co3O4/GF as anode is likely due to the inefficient ion transportation and a weak interfacial interaction between Co3O4 and graphene.22 When Co3O4/C/graphene nanocomposite was used as the anode for LIBs, the coulombic efficiency (CE) reached 85% during the first cycle, because the formation of the solid electrolyte interface (SEI) films consumed a certain amount of lithium ions during the discharge process. The partial decomposition of SEI films and the reaction of oxygen-containing functional groups on the unfolded graphene sheets with lithium ions were believed to be the primary reasons for the loss of irreversible capacity.23 Remarkably, the average CE quickly increased to 95% at the second cycle and remained close to 100% thereafter, which is due in large part to the formation of a stable SEI films on the composite electrode, further confirming that the hybrid has good reversibility of the conversion reaction between Co3O4 and Co.24 The cyclic voltammetry (CV) results (Fig. S6, ESI†) also indicate the electrochemical reversibility of Co3O4/C/graphene nanocomposite is gradually built up after the initial cycle.
 | ||
| Fig. 4 (a) Electrochemical cycling performance and the coulombic efficiency of Co3O4/C/graphene nanocomposite electrode, pure spinel Co3O4 nanoparticles and GF cycled at current density of 100 mA g−1 for 40 cycles over the potential window of 0.01–3.0 V versus Li+/Li. (b) Rate capability of Co3O4/C/graphene nanocomposite electrode. | ||
In Fig. 4b, the cell was first cycled at a low current density of 50 mA g−1 for 10 times, where an excellent high specific capacity of 1300 mA h g−1 was determined. The capacity was as high as 1200 mA h g−1 after we have increased the current density to 200 mA g−1. Yet it faded quickly as the current density increased sequentially. Galvanostatic charge–discharge curves (Fig. S7b, ESI†) were recorded at a current density of 50, 100, 200, 300, 500, 800, 1000 and then 50 mA g−1 at room temperature in the voltage range from 0.01 to 3.0 V versus Li+/Li. Apparently, the charge potential increased and the discharge potential decreased with the increase of current density, allowing higher over potential. The resulting electrode polarization is the leading cause to reduce the capacity. More importantly, the capacity recovered to 1400 mA h g−1 when the charge–discharge current density was brought back to 50 mA g−1, indicating the improved adaptability of Co3O4/C/graphene anodes. The good rate capability and cycling stability are partially attributed to the carbon coatings on Co3O4 nanoparticles. In the charging and discharging processes of LIBs, they could serve as perfect barriers to maintain their high capacities by effectively protecting the inner active materials from volume expansion and structure pulverization.4,25
Based on the above electrochemical performance, we can conclude that Co3O4/C/graphene anode materials prepared by our method have the following advantages: firstly, the “soft” interface between carbon materials and metal oxides greatly increase the capacity of LIBs. Secondly, a conductive network could not only afford more facile access of the electrolyte to anode materials, which is built up by the unfolded graphene sheets, but also contribute to the diffusion of lithium ions.8,26 EIS technique is employed to investigate the charge transfer and lithium ions diffusion kinetics thoroughly (Fig. S8, ESI†).27 The intimate proximity between graphene and Co3O4 nanoparticles coated by carbon provides superior transport pathways, enables lithium ions to intercalate and de-intercalate rapidly and freely.28
Conclusions
In summary, binder-free slurry of Co3O4/C/graphene nanocomposite with “soft” interfaces have been successfully prepared via a facile and simple calcination method, and applied as superior anode materials for giant-performance LIBs. The resulting Co3O4/C/graphene nanocomposite exhibits greatly improved electrochemical performance, including higher specific capacity (1400 mA h g−1 at a current density of 50 mA g−1), superior rate capability and good cycling stability (1050 mA h g−1 at a current density of 100 mA g−1 after 40 cycles). This excellent electrochemical property benefits from the multilayer architecture of GF and core–shell structure of Co3O4/C nanoparticles along with an interdigitated interface. The unique structure provides a large electrode/electrolyte contact area, short path length for lithium ions transport, thus sufficiently prevent volume expansion and aggregation of Co3O4 nanoparticles. Our results can pave the way to develop novel anode materials for high-performance LIBs.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 51173139, 51072151) and the China Postdoctoral Science Foundation (No. 2012M 521481).Notes and references
- (a) C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22(8), E28–E62 CrossRef CAS PubMed; (b) B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334(6058), 928–935 CrossRef CAS PubMed; (c) J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22(3), 587–603 CrossRef CAS.
- (a) J. Jiang, Y. Li, J. Liu, X. Huang, C. Yuan and X. W. Lou, Adv. Mater., 2012, 24(38), 5166–5180 CrossRef CAS PubMed; (b) P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407(6803), 496–499 CrossRef CAS PubMed; (c) X. Y. Yao, X. Xin, Y. M. Zhang, J. Wang, Z. P. Liu and X. X. Xu, J. Alloys Compd., 2012, 521, 95–100 CrossRef CAS PubMed; (d) Z. S. Wu, G. M. Zhou, L. C. Yin, W. C. Ren, F. Li and H. M. Cheng, Nano Energy, 2012, 1(1), 107–131 CrossRef CAS PubMed.
- (a) L. L. Xing, Z. H. Chen and X. Y. Xue, Solid State Sci., 2014, 32, 88–93 CrossRef CAS PubMed; (b) X. W. Lou, D. Deng, J. Y. Lee, J. Feng and L. A. Archer, Adv. Mater., 2008, 20(2), 258–262 CrossRef CAS PubMed; (c) L. Q. Tao, J. T. Zai, K. X. Wang, H. J. Zhang, M. Xu, J. Shen, Y. Z. Su and X. F. Qian, J. Power Sources, 2012, 202, 230–235 CrossRef CAS PubMed; (d) Z. S. Wu, W. C. Ren, L. Wen, L. B. Gao, J. P. Zhao, Z. P. Chen, G. M. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 4(6), 3187–3194 CrossRef CAS PubMed; (e) J. M. Xu, J. S. Wu, L. L. Luo, X. Q. Chen, H. B. Qin, V. Dravid, S. B. Mi and C. L. Jia, J. Power Sources, 2015, 274, 816–822 CrossRef CAS PubMed; (f) X. W. Lou, D. Deng, J. Y. Lee and L. A. Archer, J. Mater. Chem., 2008, 18(37), 4397 RSC.
- W. M. Zhang, X. L. Wu, J. S. Hu, Y. G. Guo and L. J. Wan, Adv. Funct. Mater., 2008, 18(24), 3941–3946 CrossRef CAS PubMed.
- (a) Y. G. Guo, Y. S. Hu, W. Sigle and J. Maier, Adv. Mater., 2007, 19(16), 2087–2091 CrossRef CAS PubMed; (b) J. C. Liu, Y. J. Xu, X. J. Ma, J. K. Feng, Y. T. Qian and S. L. Xiong, Nano Energy, 2014, 7, 52–62 CrossRef CAS PubMed.
- (a) H. Wang, Y. Yang, Y. Liang, J. T. Robinson, Y. Li, A. Jackson, Y. Cui and H. Dai, Nano Lett., 2011, 11(7), 2644–2647 CrossRef CAS PubMed; (b) H. P. Jia, P. F. Gao, J. Yang, J. L. Wang, Y. N. Nuli and Z. Yang, Adv. Energy Mater., 2011, 1(6), 1036–1039 CrossRef CAS PubMed; (c) R. Mo, Z. Lei, K. Sun and D. Rooney, Adv. Mater., 2014, 26(13), 2084–2088 CrossRef CAS PubMed.
- (a) H. Gwon, H. S. Kim, K. U. Lee, D. H. Seo, Y. C. Park, Y. S. Lee, B. T. Ahn and K. Kang, Energy Environ. Sci., 2011, 4(4), 1277 RSC; (b) D. H. Wang, D. W. Choi, J. Li, Z. G. Yang, Z. M. Nie, R. Kou, D. H. Hu, C. M. Wang, L. V. Saraf, J. G. Zhang, I. A. Aksay and J. Liu, ACS Nano, 2009, 3(4), 907–914 CrossRef CAS PubMed; (c) G. X. Wang, X. P. Shen, J. Yao and J. Park, Carbon, 2009, 47(8), 2049–2053 CrossRef CAS PubMed; (d) X. C. Chen, W. Wei, W. Lv, F. Y. Su, Y. B. He, B. Li, F. Kang and Q. H. Yang, Chem. Commun., 2012, 48(47), 5904–5906 RSC.
- M. Zhang, E. Uchaker, S. Hu, Q. Zhang, T. Wang, G. Cao and J. Li, Nanoscale, 2013, 5(24), 12342–12349 RSC.
- (a) R. Wang, C. Xu, J. Sun, Y. Liu, L. Gao and C. Lin, Nanoscale, 2013, 5(15), 6960–6967 RSC; (b) L. Noerochim, J. Z. Wang, S. L. Chou, D. Wexler and H. K. Liu, Carbon, 2012, 50(3), 1289–1297 CrossRef CAS PubMed; (c) I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334(6052), 75–79 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80(6), 1339 CrossRef CAS.
- Q. Li, L. Dong, Y. Liu, H. Xie and C. Xiong, Carbon, 2011, 49(3), 1047–1051 CrossRef CAS PubMed.
- Y. Lei, C. Xiong, L. Dong, H. Guo, X. Su, J. Yao, Y. You, D. Tian and X. Shang, Small, 2007, 3(11), 1889–1893 CrossRef CAS PubMed.
- (a) J. M. Xu, J. S. Wu, L. L. Luo, X. Q. Chen, H. B. Qin, V. Dravid, S. B. Mi and C. L. Jia, J. Power Sources, 2015, 274, 816–822 CrossRef CAS PubMed; (b) L. Pan, H. Zhao, W. Shen, X. Dong and J. Xu, J. Mater. Chem. A, 2013, 1(24), 7159 RSC.
- (a) R. Rodriguez, R. Herrera, L. A. Archer and E. P. Giannelis, Adv. Mater., 2008, 20(22), 4353–4358 CrossRef CAS PubMed; (b) Q. Li, L. Dong, F. Sun, J. Huang, H. Xie and C. Xiong, Chem.–Eur. J., 2012, 18(23), 7055–7059 CrossRef CAS PubMed; (c) A. B. Bourlinos, V. Georgakilas, V. Tzitzios, N. Boukos, R. Herrera and E. P. Giannelis, Small, 2006, 2(10), 1188–1191 CrossRef CAS PubMed; (d) A. B. Bourlinos, S. R. Chowdhury, R. Herrera, D. D. Jiang, Q. Zhang, L. A. Archer and E. P. Giannelis, Adv. Funct. Mater., 2005, 15(8), 1285–1290 CrossRef CAS PubMed.
- (a) Y. S. Hu, Y. G. Guo, R. Dominko, M. Gaberscek, J. Jamnik and J. Maier, Adv. Mater., 2007, 19(15), 1963–1966 CrossRef CAS PubMed; (b) H. X. Ji, X. L. Wu, L. Z. Fan, C. Krien, I. Fiering, Y. G. Guo, Y. Mei and O. G. Schmidt, Adv. Mater., 2010, 22(41), 4591–4595 CrossRef CAS PubMed.
- Y. C. Si and E. T. Samulski, Chem. Mater., 2008, 20(21), 6792–6797 CrossRef CAS.
- (a) X. L. Li, Y. Q. Liu, L. Fu, L. C. Cao, D. C. Wei and Y. Wang, Adv. Funct. Mater., 2006, 16(18), 2431–2437 CrossRef CAS PubMed; (b) M. Terrones, W. K. Hsu, A. Schilder, H. Terrones, N. Grobert, J. P. Hare, Y. Q. Zhu, M. Schwoerer, K. Prassides, H. W. Kroto and D. R. M. Walton, Appl. Phys. A: Mater. Sci. Process., 1998, 66, 307–318 CrossRef CAS; (c) J. H. Baek, J. Y. Park, A. R. Hwang and Y. C. Kang, Bull. Korean Chem. Soc., 2012, 33(4), 1242–1246 CrossRef CAS; (d) G. X. Wang, J. Yang, J. Park, X. L. Gou, B. Wang, H. Liu and J. Yao, J. Phys. Chem. C, 2008, 112(22), 8192–8195 CrossRef CAS.
- M. B. Zheng, J. Cao, S. T. Liao, J. S. Liu, H. Q. Chen, Y. Zhao, W. J. Dai, G. B. Ji, J. M. Cao and J. Tao, J. Phys. Chem. C, 2009, 113(9), 3887–3894 CAS.
- (a) N. Xue, W. Du, A. Gupta, W. Shyy, A. Marie Sastry and J. R. R. A. Martins, J. Electrochem. Soc., 2013, 160(8), A1071–A1078 CrossRef CAS PubMed; (b) C. B. Wang, C. W. Tang, S. J. Gau and S. H. Chien, Catal. Lett., 2005, 101(1–2), 59–63 CAS.
- (a) Y. Wang, Y. Y. Shao, D. W. Matson, J. H. Li and Y. H. Lin, ACS Nano, 2010, 4(4), 1790–1798 CrossRef CAS PubMed; (b) X.-L. Xu, Z.-H. Chen, Y. Li, W.-K. Chen and J.-Q. Li, Surf. Sci., 2009, 603(4), 653–658 CrossRef CAS PubMed; (c) H. A. E. Hagelin-Weaver, G. B. Hoflund, D. M. Minahan and G. N. Salaita, Appl. Surf. Sci., 2004, 235(4), 420–448 CrossRef CAS PubMed.
- (a) L. Pan, H. Zhao, W. Shen, X. Dong and J. Xu, J. Mater. Chem. A, 2013, 1(24), 7159 RSC; (b) T. He, D. Chen, X. Jiao and Y. Wang, Adv. Mater., 2006, 18(8), 1078–1082 CrossRef CAS PubMed; (c) C. Yuan, L. Yang, L. Hou, J. Li, Y. Sun, X. Zhang, L. Shen, X. Lu, S. Xiong and X. W. D. Lou, Adv. Funct. Mater., 2012, 22(12), 2560–2566 CrossRef CAS PubMed.
- (a) X. C. Chen, Chem. Commun., 2012, 48(47), 5904–5906 RSC; (b) H. Gwon, H.-S. Kim, K. U. Lee, D.-H. Seo, Y. C. Park, Y.-S. Lee, B. T. Ahn and K. Kang, Energy Environ. Sci., 2011, 4(4), 1277 RSC; (c) Z. Li, Y. Mi, X. Liu, S. Liu, S. Yang and J. Wang, J. Mater. Chem., 2011, 21(38), 14706 RSC; (d) L. Noerochim, J.-Z. Wang, S.-L. Chou, D. Wexler and H.-K. Liu, Carbon, 2012, 50(3), 1289–1297 CrossRef CAS PubMed.
- C. Ban, Z. Wu, D. T. Gillaspie, L. Chen, Y. Yan, J. L. Blackburn and A. C. Dillon, Adv. Mater., 2010, 22(20), E145–E149 CrossRef CAS PubMed.
- H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 Search PubMed.
- (a) W. M. Zhang, X. L. Wu, J. S. Hu, Y. G. Guo and L. J. Wan, Adv. Funct. Mater., 2008, 18(24), 3941–3946 CrossRef CAS PubMed; (b) W. M. Zhang, J. S. Hu, Y. G. Guo, S. F. Zheng, L. S. Zhong, W. G. Song and L. J. Wan, Adv. Mater., 2008, 20(6), 1160–1165 CrossRef CAS PubMed; (c) G. Cui, Y. S. Hu, L. Zhi, D. Wu, I. Lieberwirth, J. Maier and K. Mullen, Small, 2007, 3(12), 2066–2069 CrossRef CAS PubMed; (d) W. M. Zhang, J. S. Hu, Y. G. Guo, S. F. Zheng, L. S. Zhong, W. G. Song and L. J. Wan, Adv. Mater., 2008, 20(6), 1160–1165 CrossRef CAS PubMed.
- L. Zou, L. Gan, F. Kang, M. Wang, W. Shen and Z. Huang, J. Power Sources, 2010, 195(4), 1216–1220 CrossRef CAS PubMed.
- (a) Q. C. Zhuang, T. Wei, L. L. Du, Y. L. Cui, L. Fang and S. G. Sun, J. Phys. Chem. C, 2010, 114(18), 8614–8621 CrossRef CAS; (b) D. Fang, L. Li, W. Xu, G. Li, G. Li, N. Wang, Z. Luo, J. Xu, L. Liu, C. Huang, C. Liang and Y. Ji, J. Mater. Chem. A, 2013, 1(42), 13203 RSC.
- S. L. Candelaria, Y. Shao, W. Zhou, X. Li, J. Xiao, J.-G. Zhang, Y. Wang, J. Liu, J. Li and G. Cao, Nano Energy, 2012, 1(2), 195–220 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra11104h |
| This journal is © The Royal Society of Chemistry 2015 |