
An amperometric immunosensor for detection of Streptococcus suis serotype 2 using a nickel–gold nanocomposite as a tracer matrix
Qiang
Zhu
ab,
Yaqin
Chai
*a,
Ying
Zhuo
a and
Ruo
Yuan
*a
aEducation Ministry Key Laboratory on Luminescence and Real-Time Analysis, College of Chemistry and Chemical Engineering, Southwest University, Chongqing 400715, PR China. E-mail: yqchai@swu.edu.cn; yuanruo@swu.edu.cn; Fax: +86-23-68252277; Tel: +86-23-68253172
bKey Laboratory of Green Synthesis and Applications, College of Chemistry, Chongqing Normal University, Chongqing 401331, China
First published on 7th September 2015
Abstract
A simple electrochemical immunosensor for determination of Streptococcus suis serotype 2 (SS2) is presented. Nickel hexacyanoferrate nanoparticles (NiHCFNPs) and gold nanocages (AuNCs) were combined together through complexation between the cyanide (CN−) and gold ions, and acted as a carrier for immobilization of the detection antibody (Ab2). The capture antibody (Ab1) was combined by a protein A (PA) and gold nanoparticle (AuNP) modified electrode. In a sandwich-type immunoassay mode, ascorbic acid (AA) was added to the base solution and its oxidation was catalyzed by the AuNCs–NiHCFNPs complex to greatly amplify the response signal. The resulting immunosensor exhibited excellent selectivity and sensitivity, with a linear concentration range of 0.0005–80 ng mL−1 and a detection limit of 0.15 pg mL−1.
1. Introduction
The first case of Streptococcus suis (S. suis) infection was reported in 1954,1 and fourteen years later, the first human S. suis case was discovered in Denmark. Subsequently, other cases were reported in other northern European countries and Hong Kong.2–4 In July 2005, a sudden outbreak of human S. suis infection in the Sichuan Province of China sparked a nationwide food scare; 38 people died and nearly all the pigs in the district were slaughtered and deep-buried. Researchers have proven that there were at least 35 serotypes of S. suis with varying virulence,5 and Streptococcus suis type 2 (SS2) was regarded as the chief pathogen for human and animal infection. The fatality rate of SS2 was about 7%; nearly half of the patients suffered from the sequelae of vertigo and hearing loss.To date, various analytical methods have been developed for the detection of SS2, such as routine biochemistry assays, agar gel precipitation, enzyme-linked immunosorbent assay (ELISA),6 immunochromatographic assay,7 and polymerase chain reaction (PCR).8 However, most of these methods are time-consuming, costly and need laborious procedures. Electrochemical sensors are attractive for their advantages of simple operation and high sensitivity and specificity, and they can be used to detect various target proteins.9,10 However, there have rarely been reports of electrochemical determination of SS2.11,12 Here, we attempted to develop a simple, quick and sensitive electrochemical immunosensor for detection of SS2.
Metal hexacyanoferrates (MHCFs) have been very active in electrochromism, ion exchange and electrocatalysis. For example, copper hexacyanoferrate (CuHCF), manganese hexacyanoferrate (MnHCF), and nickel hexacyanoferrate (NiHCF) were effective catalysts for ascorbic acid (AA) oxidation.13 NiHCF has been used as a redox probe in various biosensors.14–16 As the CN− in NiHCF could easily bind with a gold ion, this characteristic could be used in the preparation of a gold–nickel nanocomposite by means of mixing.17
Since gold colloid was reported by Faraday in 1857,18 various noble-metal nanostructures have been the focus of much research; they have shown unique properties in catalysis, photonics, electronics, and information storage.19–23 Gold nanomaterials, with good biocompatibility, excellent conductivity and a large specific surface area, have been used for biomedical applications including drug delivery, tissue/tumor imaging and photothermal therapy.24–27 Gold nanocages (AuNCs) have extremely thin but electrically continuous walls, which could provide a sufficiently large surface area to accommodate both the oxidation and reduction half reactions, and they may be more active than similar-sized non-hollow nanoparticles.28 Here, AuNCs were prepared and bonded with NiHCFNPs to form a AuNCs–NiHCFNPs (or Au–NiNPs for short) complex as the Ab2 substrate.
To construct the biosensor, gold nanoparticles (AuNPs) were electrochemically deposited on the surface of a glass carbon electrode (GCE), then protein A (PA) was immobilized on the AuNPs as a linkage reagent to integrate Ab1. The Au–NiNPs complex was used to combine Ab2 as a tracer, which not only provided a redox peak for detection, but also performed catalysis of AA oxidation to amplify the response signal. The resulting immunosensor exhibited excellent sensitivity as expected. The features of the nano-materials and the fabrication process of the sensor were characterized by scanning electron microscopy (SEM) and electrochemical methods. All kinds of experimental parameters such as the optimal concentration of AA, temperature, pH, selectivity, stability and preliminary applications were discussed.
2. Experimental section
2.1. Reagents and apparatus
Streptococcus agalactiae (1.B.501) sc-73072 lot # J1408 (Ab1), Streptococcus group B (072) sc-58045 lot # B1607 (Ab2) and Streptococcus suis 2 lot # A2209 (SS2) were obtained from Santa Cruz Biotechnology Inc. (USA). Protein A, gold chloride tetrahydrate (HAuCl4) and L-ascorbic acid (AA) were obtained from Sigma Chemical Co. (St. Louis., MO. USA). Gold nanoparticles with a mean size of 16 nm were prepared by reducing gold chloride tetrahydrate with sodium citrate. Serum specimens were provided by Daping Hospital of Third Military Medical University (Chongqing, China), the study protocols were approved by the local Institutional Review Board (IRB), and informed consent was obtained from the patients. All of the chemicals used were of analytical grade and solutions were prepared using ultrapure water (specific resistance of 18 MΩ cm−1).Differential pulse voltammetry (DPV) and cyclic voltammetric (CV) tests were performed with a CHI 660C electrochemical workstation (Shanghai Chenhua Instrument, China). The three electrode electrochemical system contained a modified glass carbon electrode (GCE, Φ = 4 mm) as the working electrode, a platinum wire as the counter electrode and a saturated calomel electrode (SCE) as the reference electrode. Ultraviolet-visible absorption spectra were recorded using a 2450 UV-vis spectrometer with a 1.00 cm quartz cuvette in the range of 200–800 nm (Shimadzu, Japan). Scanning electron microscopy (SEM, S-4800, Hitachi, Japan) was employed to estimate the morphology of the prepared nanomaterials. X-ray photoelectron spectroscopy (XPS) analysis was measured with a VG Scientific ESCALAB 250 spectrometer operating with Al Kα X-ray (1486.6 eV) as the light source.
2.2. Preparation of gold nanocages (AuNCs)
The procedure included two steps: preparation of silver nanocubes and using the Ag template to reduce the HAuCl4 solution to generate the Au nanostructures:28| 3Ag(s) + [AuCl4]−(aq) → Au(s) + 3Ag+(aq) + 4Cl−(aq) |
Ag nanocubes were prepared by the following steps: 30 mL of ethylene glycol (EG) was heated to 150 °C using an oil bath, then 10 mL of EG solution containing 0.2 g of polyvinylpyrrolidone (PVP) was added with magnetic stirring, and stirring was maintained for 5 min. Next, 400 µL of EG containing 3 mM Na2S was added dropwise, and following this step, 3 mL of EG containing 282 mM AgNO3 was added to the solution dropwise, and the color of the solution began to change from white to brown. The resulting Ag nanocubes were collected by centrifugation and washing with acetone several times (SEM morphology is shown in Fig. 1B).
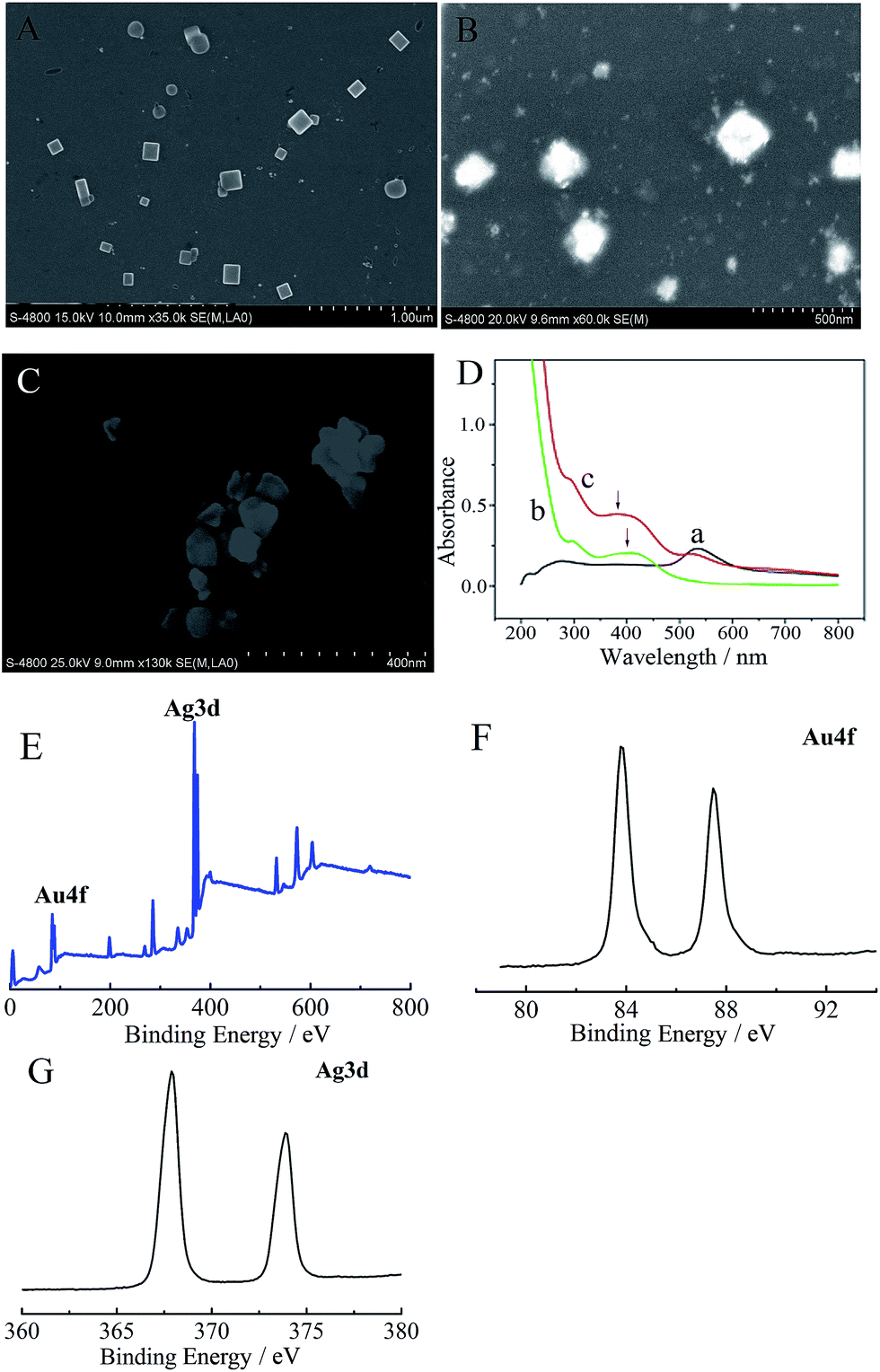 | ||
| Fig. 1 Characteristics of the prepared nanomaterials. The SEM morphology of the Au nanocage (A), Ag nanocube (B) and Au–NiNPs complex (C); (D) UV-vis spectrum of the Au–NiNPs solution: NiNPs (a), AuNCs (b) and Au–NiNPs (c); XPS analysis of the Au nanocages: the full region of XPS (E), Au4f region (F) and Ag3d region (G). | ||
Whereafter, the resulted Ag nanocubes were redispersed in 30 mL of water and heated to boil for 10 min, then 100 µL of HAuCl4 solution (10 mg mL−1) was slowly added and the reflux maintained for 30 min. The color of the solution changed from brown to pink. Finally, the achieved AuNCs were collected and redispersed in water for use. The SEM morphology of the AuNCs is shown in Fig. 1A; the edge length is approximately 90 nm.
X-ray photoelectron spectroscopy (XPS) was used to analyze the elemental makeup of the AuNCs. The XPS signature of the Au4f doublet (83.4 eV and 87.5 eV for the 4f7/2 and 4f5/2 regions, respectively) and Ag3d doublet (368.5 eV and 373.4 eV for the 3d3/2 and 3d5/2 regions, respectively) were observed (Fig. 1E–G), indicating that the AuNCs were a kind of alloy of gold and silver.
2.3. Preparation of NiHCFNPs
According to a typical method in the literature,12 30 mL of a solution containing 0.012 M NiCl2 was added to 30 mL of 0.05 mol L−1 K3Fe(CN)6 solution containing 0.05 M KCl with vigorous magnetic stirring, then constantly stirred for 10 min, and the resulting solution was centrifugated, washed and redispersed in 10 mL of water for use. The size of the nano-spheres was about 40 nm (measured with SEM under dry conditions).2.4. Preparation of the Ab2 composite
To prepare the Au–NiNPs complex, 1.0 mL of NiNPs solution was added to 1 mL of AuNCs solution under vigorous stirring for 8 h. Through this procedure, the Au–NiNPs complex was spontaneously generated. After centrifugal separation, the complex was redispersed in water for use (the SEM morphology of Au–NiNPs is shown in Fig. 1C).The UV-vis spectrum of Au–NiNPs is presented in Fig. 1D. The maximum absorption peak of the AuNCs is located at 540 nm (curve a); NiNPs exhibited a strong absorbance at 290 nm and a broad absorption peak at around 405 nm (curve b); in the Au–NiNPs solution, the absorbance peak of the NiNPs presented an obvious blue shift, the absorbance peak of the AuNCs became less pronounced and the background absorbance obviously increased (curve c), which can be ascribed to the interaction between the AuNCs and NiNPs.
Next, 200 µL of 100 ng mL−1 Ab2 solution was added to 1.0 mL of Au–NiNPs complex solution, and gently stirred for 12 h at 4 °C to form the Ab2/Au–NiNPs composite, after which 200 µL of BSA (1 mg mL−1) was added to block the nonspecific adsorption sites, and the resulting composite was centrifugated and washed to remove excess reagent, and redispersed in 1 mL of water for use.
2.5. Fabrication procedure for the immunosensor
Preliminarily, the glassy carbon electrode (GCE, Φ = 4 mm) was treated with 0.3 and 0.05 µm alumina slurry and ultrasonic washing, then the GCE was put in 2 mL of 1 mM HAuCl4 solution and a constant potential of −0.2 V was applied for 30 s for the fabrication of AuNPs on the electrode surface. Next, the electrode was immersed in 0.2 mg mL−1 PA (pH 3) aqueous solution for 2 h to combine PA through electrostatic adsorption, then incubated with 100 ng mL−1 Ab1 solution for 12 h at 4 °C, and finally rinsed with water and stored at 4 °C for use. Each procedure was followed by careful washing.2.6. The detection principle of the immunosensor
Based on a sandwich-type immunoassay format, the immunosensor was first incubated with the target antigen for 40 min at 37 °C, then incubated with 20 µL of Ab2 composite for 40 min at 37 °C, and acted as the working electrode and was detected by DPV scan in 0.1 M PBS (pH 7.0) containing 5 mM AA. The following catalysis reaction between the Ab2 composite and AA would happen (Fig. 2).
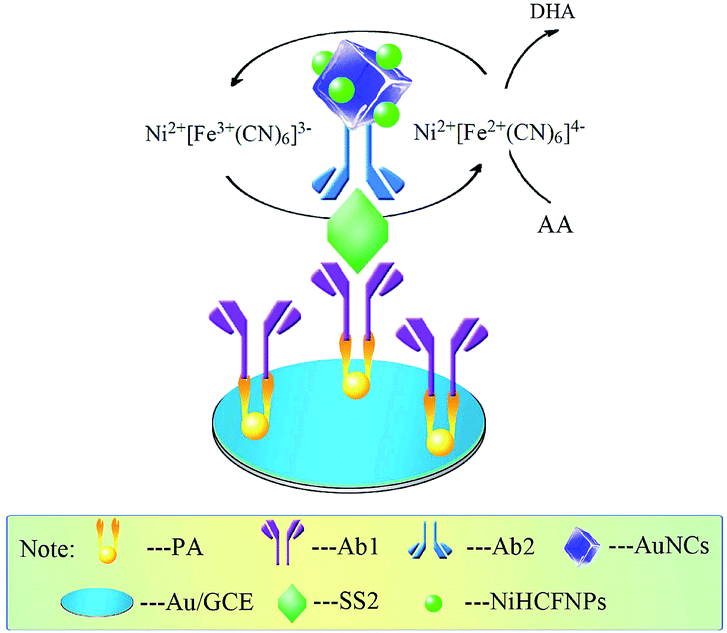 | ||
| Fig. 2 The detection principle of the immunosensor. | ||
With this reaction, the response current could be greatly amplified, and the current intensity was quantitatively related to the content of target antigen.
3. Results and discussion
3.1. CV characterization of the fabrication procedure of the immunosensor
The CV characterization of the fabrication procedure in 5.0 mM K3[Fe(CN)6]/K4[Fe(CN)6] solution is presented in Fig. 3. Typical reversible redox peaks of ferricyanide ions could be observed for the bare GCE (Fig. 3A, curve a); after electrodeposition of AuNPs on the surface of the GCE, the redox peaks obviously increased (Fig. 3A, curve b) due to the excellent conductivity of the AuNPs. When combined with PA, the peak current significantly decreased (Fig. 3A, curve c), which could be ascribed to the poor conductivity of the protein. In successive steps of immobilization of Ab1 and incubation of SS2, the peak currents both decreased (Fig. 3A, curve d and e) for the same reason. However, an increased peak current was observed after the incubation of the Ab2 composite (Fig. 3A, curve f), which could be attributed to the Au–NiNPs complex which could accelerate the electron transfer. When the immunosensor was placed in 0.1 M PBS (pH 7.0) containing 5 mM AA, no obvious peak was observed (Fig. 3B, curve a). However, after the immunosensor was incubated with the Ab2 composite, a sharp amplification of the peak could be achieved (Fig. 3B, curve b), which proved that the Ab2 composite was successfully immobilized, and could strongly amplify the response signal. Based on the above analysis, we concluded that the fabrication procedures were practicable. | ||
| Fig. 3 (A) CV curves of the fabrication procedures of the immunosensor in 5 mM K3[Fe(CN)6]/K4[Fe(CN)6] solution: (a) bare GCE; (b) Au/GCE; (c) PA/Au/GCE; (d) Ab1/PA/Au/GCE; (e) SS2/Ab1/PA/Au/GCE; (f) Ab2/SS2/Ab1/PA/Au/GCE. (B) CV curves of the immunosensor before (a) and after (b) incubation of the Ab2 composite (in pH 7.0 PBS containing 5 mM AA). | ||
3.2. Optimization of test conditions
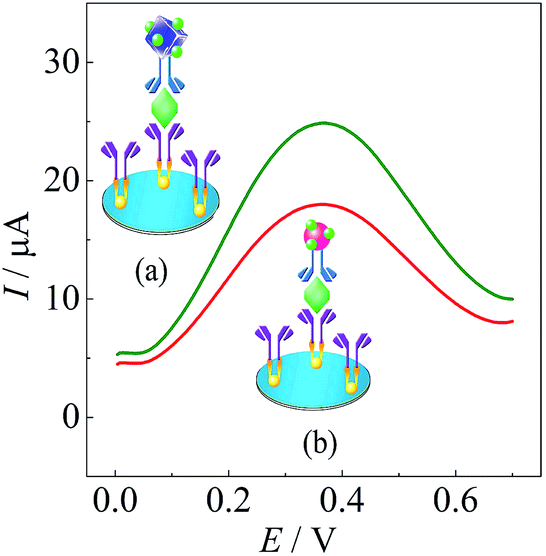 | ||
| Fig. 4 Investigation of the response signal of (a) AuNC and (b) AuNP based immunosensors. | ||
3.3. Performance of the immunosensor
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logc + 17.65 (ng mL−1), R2 = 0.9900 (Fig. 5B).
logc + 17.65 (ng mL−1), R2 = 0.9900 (Fig. 5B).
 | ||
| Fig. 5 The DPV responses of the immunosensor after incubation with standard concentrations of SS2 under optimal conditions. | ||
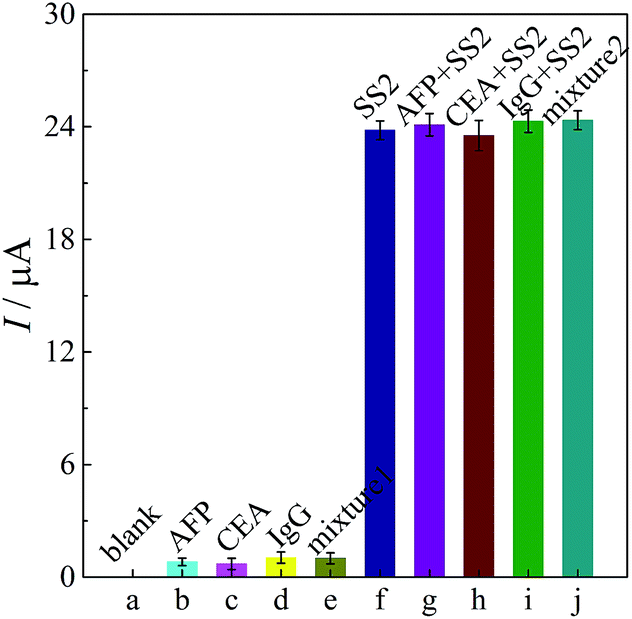 | ||
| Fig. 6 Specificity test of the immunosensor. (a) 0 ng mL−1 SS2, (b) 100 ng mL−1 AFP, (c) 80 ng mL−1 CEA, (d) 100 ng mL−1 IgG, (e) a mixture of the three interference proteins, (f) 10 ng mL−1 SS2, (g) 100 ng mL−1 AFP + 10 ng mL−1 SS2, (h) 80 ng mL−1 CEA + 10 ng mL−1 SS2 CEA, (i) 100 ng mL−1 IgG + 10 ng mL−1 SS2, (j) a mixture of the three interference proteins + 10 ng mL−1 SS2. | ||
Long-term storage stability was also examined. When the immunosensors were stored in a refrigerator for a week, the average decrease in the value of the peak current was less than 4.6% compared to the freshly prepared immunosensor, and this value became 9.3% in two weeks. Therefore, the stability of the proposed immunosensor was acceptable.
3.4. Recovery test
Recovery experiments were performed by standard addition methods in human serum. The serum samples were diluted to a suitable concentration with PBS (pH 7.0). The experimental results are shown in Table 1; the recovery was in the range of 97.0–104.0%. This data was acceptable, which proved that the immunosensor could perform the determination of SS2 in real samples.4. Conclusion
For quick and sensitive detection of SS2, a sandwich-type amperometric immunosensor was developed. AuNCs and NiHCFNPs were bound together through simple complexation and acted as an Ab2 substrate and signal probe, as well as a catalytic reagent for AA oxidation to amplify the response signal. The resulting immunosensor was very simple, sensitive and low cost, and could provide a promising method for the detection of SS2 in clinical diagnosis.Acknowledgements
The authors would like to thank the NNSF of China (51473136 and 21275119), Ministry of Education of China (Project 708073), Fundamental Research Funds for the Central Universities (XDJK2015A002, XDJK2014A012, XDJK2013A008). Scientific and Technological Project of CQEC (KJ130640). Program for Innovation Team Building at Institutions of Higher Education in Chongqing (KJTD201309).References
- H.-I. Field, D. Buntain and J.-T. Done, Vet. Rec., 1954, 66, 453–455 Search PubMed.
- B. Perch, P. Kristjansen and K. Skadhauge, Acta Pathol. Microbiol. Scand., 1968, 74, 69–76 CrossRef CAS PubMed.
- J.-P. Arends and H.-C. Zanen, Rev. Infect. Dis., 1988, 10, 131–137 CrossRef CAS PubMed.
- B.-F. McLendon, A.-J. Bron, C.-J. Mitchell and L.-R. Infirmary, Br. J. Ophthalmol., 1978, 62, 729–731 CrossRef CAS.
- H.-J. Yu, H.-Q. Jing, Z.-H. Chen, H. Zheng, X.-P. Zhu, H. Wang, S.-W. Wang, L.-G. Liu, R.-Q. Zu, L.-Z. Luo, N.-J. Xiang, H.-L. Liu, X.-C. Liu, Y.-L. Shu, S.-S. Lee, S.-K. Chuang, Y. Wang and J.-G. Xu, Emerging Infect. Dis., 2006, 6, 914–920 CrossRef PubMed.
- E. Martin del Campo Sepúlveda, E. Altman, M. Kobisch, S.-D. Allaire and M. Gottschalk, Vet. Microbiol., 1996, 1, 113–125 CrossRef.
- Y. Ju, H.-J. Hao, G.-H. Xiong, H.-R. Geng, Y.-L. Zheng, J. Wang, Y. Cao, Y.-H. Yang, X.-H. Cai and Y.-Q. Jiang, Vet. Immunol. Immunopathol., 2010, 2, 207–211 CrossRef PubMed.
- H.-J. Wisselink, F.-H. Reek, U. Vecht, N. Stockhofe-Zurwieden, M.-A. Smits and H.-E. Smith, Vet. Microbiol., 1999, 2, 143–157 CrossRef.
- J.-M. Hu, Y.-J. Yu, J.-C. Brooks, L.-A. Godwin, S. Somasundaram, F. Torabinejad, J. Kim, C. Shannon and C.-J. Easley, J. Am. Chem. Soc., 2014, 136, 8467–8474 CrossRef CAS PubMed.
- J.-M. Hu, T. Wang, J. Kim, C. Shannon and C.-J. Easley, J. Am. Chem. Soc., 2012, 134, 7066–7072 CrossRef CAS PubMed.
- H.-J. Wang, R. Yuan, Y.-Q. Chai, Y.-L. Cao, X.-X. Gan, Y. Chen and Y. Wang, Biosens. Bioelectron., 2013, 43, 63–68 CrossRef CAS PubMed.
- Q. Zhu, Y.-Q. Chai, R. Yuan, Y. Zhuo, J. Han, Y. Li and N. Liao, Biosens. Bioelectron., 2013, 43, 440–445 CrossRef CAS PubMed.
- T.-H. Tsai, T.-W. Chen, S.-M. Chen and R. Sarawathi, Russ. J. Electrochem., 2012, 3, 291–301 CrossRef.
- M.-H. Yang, J.-H. Jiang, Y.-H. Yang, F.-L. Qu, Y.-S. Lu and G.-L. Shen, Anal. Chim. Acta, 2006, 571, 211–217 CrossRef CAS PubMed.
- P. Prabhu, R.-S. Babu and S.-S. Narayanan, Sens. Actuators, B, 2011, 156, 606–614 CrossRef CAS PubMed.
- X.-Y. Wang, Y. Zhang, C.-E. Banks, Q.-Y. Chen and X.-B. Ji, Colloids Surf., B, 2010, 78, 363–366 CrossRef CAS PubMed.
- J. Han, Y. Zhuo, Y.-Q. Chai, Y.-L. Yuan and R. Yuan, Biosens. Bioelectron., 2012, 31, 399–405 CrossRef CAS PubMed.
- M. Faraday, Philos. Trans. R. Soc. London, 1857, 147, 145–181 CrossRef.
- S.-K. Ghosh and T. Pal, Chem. Rev., 2007, 11, 4797–4862 CrossRef PubMed.
- J.-S. Lee, P.-A. Ulmann, M.-S. Han and C.-A. Mirkin, Nano Lett., 2008, 2, 529–533 CrossRef PubMed.
- L.-R. Hirsch, R.-J. Stafford, J.-A. Bankson, B.-R. Sershen, R.-E. Price, J.-D. Hazle, N.-J. Halas and J.-L. West, Inorg. Chem., 2003, 19, 7544–7554 Search PubMed.
- J. Zeng, J.-L. Huang, W. Lu, X.-P. Wang, B. Wang, S.-Y. Zhang and J.-G. Hou, Adv. Mater., 2007, 19, 2172–2176 CrossRef CAS PubMed.
- J.-Y. Chen, B. Wiley, Z.-Y. Li, D. Campbell, F. Saeki, H. Cang, L. Au, J. Lee, X. Li and Y. Xia, Adv. Mater., 2005, 17, 2255–2261 CrossRef CAS PubMed.
- Y.-G. Sun, B.-T. Mayers and Y. Xia, Nano Lett., 2002, 2, 481–485 CrossRef CAS.
- Y.-G. Sun, B.-T. Mayers and Y. Xia, Adv. Mater., 2003, 15, 641–646 CrossRef CAS PubMed.
- Y.-G. Sun and Y. Xia, Adv. Mater., 2003, 15, 695–699 CrossRef CAS PubMed.
- S.-E. Skrabalak, L. Au, X. Li and Y. Xia, Nat. Protoc., 2007, 9, 2182–2190 CrossRef PubMed.
- J. Zeng, Q. Zhang, J. Chen and Y. Xia, Nano Lett., 2009, 1, 30–35 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |