
Deuterated hydrazino-s-triazine as highly-efficient labelling reagent for glycan relative quantification analysis using electrospray ionization mass spectrometry†
Ming-Zhe Zhaoa,
Cai Tieb,
Yi-Wei Zhanga,
Yan Denga,
Fang-Ting Zhanga,
Ying-Lin Zhou*a and
Xin-Xiang Zhang*a
aBeijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Biochemistry and Molecular Engineering of Ministry of Education, Institute of Analytical Chemistry, College of Chemistry, Peking University, Beijing, 100871, China. E-mail: zhouyl@pku.edu.cn; zxx@pku.edu.cn; Fax: +86-10-62754112, +86-10-62754680; Tel: +86-10-62754112, +86-10-62754680
bState Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Peking Union Medical College & Chinese Academy of Medical Sciences, Beijing, 100050, China
First published on 14th September 2015
Abstract
Herein, an innovative stable-isotope relative quantification strategy for N-glycans was achieved using a self-designed non-reductive hydrazino-s-triazine deuterated derivative as a labelling reagent combined with mass spectrometry. As much as a 20 Da mass shift could effectively distinguish different forms of N-glycans labelled with normal and heavy hydrazino-s-triazine at the reducing end. Especially for glycans with high molecular weight, qualitative identification could be significantly simplified on account of avoiding isotopic distributions overlapping in the mass spectra. Meanwhile, a deuterium derivative with high purity guaranteed the accuracy of the relative quantification. The obviously undifferentiated response toward normal and heavy hydrazino-s-triazine labelled glycans in electrospray ionization mass spectrometry confirmed the feasibility and reliability of our proposed strategy, which was further demonstrated by relative quantification of the mixtures of labelled N-glycans cleaved from ovalbumin as a model sample. Finally, we adopted human serum as a complex sample and successfully achieved highly accurate detection of 48 N-glycans. The deuterated hydrazino-s-triazine labelling reagent exhibited a great potential to promote the development of highly-efficient, accurate and reliable N-glycan relative quantification in pharmacy and diagnosis research.
1. Introduction
Glycosylation, one of the most common and critical protein co/post-translation modifications, has been proved to play an essential role in modulating biological functions.1,2 Increasing evidence has been revealed to support the fact that glycoprotein folding, conformations, and bio-functions are regulated by glycan modifications.3 In the development of glycomics, the glycosylation process is further associated with organism disorders including serious diseases like rheumatoid arthritis,4,5 diabetes,6 inflames7 and tumors.8,9 As one of the most frequent protein glycosylation, N-linked glycans have been regarded as the most potential candidates for sensitive and specific biomarkers to fulfil both disease prediction and diagnosis. Accurate and reliable N-glycans qualification and quantification are therefore crucial for further glycosylation biomarkers discovery.Glycosylation analysis is generally complicated by its bio-diversity generated from compositions, branch structures and conformations.10–12 As a powerful structural identification and profiling tool, mass spectrometry (MS) has been widely applied in N-glycans characterization. However, the low ionization efficiency of N-glycans greatly limits the quantification of low-abundance N-glycans in biological samples. Thus, to achieve higher sensitivity, a variety of derivatization methods of glycans are developed to improve the ionization efficiency to obtain enhanced MS response, including permethylation,13,14 reductive amination,15,16 Michael addition17 and non-reductive hydrazine labelling.18,19 Illuminated by MS based relative quantification methodologies in proteomics,20 stable-isotope labelling (15N, 13C, 18O and deuterium) has been regarded as an appealing strategy in glycans quantification.21
Deuterium labelling was presented as the earliest developed22 and most frequently used methodology for N-glycan relative quantification owing to high purity and low cost of derivatization reagents. Admittedly, challenges with deuterium labelling associated with H interchange (H/D exchange reaction) always existing, by appropriate design of structures, H/D exchange reaction could be avoided. Three main labelling methods using deuterium derivative to relatively quantify N-glycans were involved as follows: (1) stable-isotope labelling via deuteriomethyl iodide (CD3I) to produce a 3 Da mass difference per functional group methylated23 or CH2DI for 1 Da mass shift,24 (2) incorporating d0-, d4-, d8- and d12-labelling reagents into N-glycans through reductive amination to obtain a mass increase of 4 Da,21,25 (3) adding isotopic phenyl-3-methyl-5-pyrazolone tag to create a mass difference of 5 Da.26 Increased mass shift to 10 Da based on two molecular addition greatly simplified the peak identification in mass spectra. Nevertheless, a chromatographic shift caused by deuterium brought about analytical variability,27 which was commonly settled by replacing the reverse phase chromatography with hydrophilic interaction chromatography (HILIC) or capillary electrophoresis (CE).
Introducing 13C into derivative glycans was another commonly adoptive N-glycan relative quantification strategy, including 12CH3I/13CH3I for earliest developed permethylation to produce a 1 Da mass increase for each reaction group,24 as well as 12C6/13C6-reductive amination reagents28–31 and 12C6/13C6-non-reductive hydrazide reagents32 to label glycan samples differentially with 6 Da mass shift. Obvious chromatographic effects were disappeared by replacing deuterium with 13C incorporation. However, the main problem lies in the expensive price of 13C derivatization reagents.
15N differentiated mass tags for glycan relative quantification were reported to be achieved by metabolic incorporation of stable isotope into the glycans of cultured cells through the hexosamine biosynthetic pathway,33 resulting in a 1 Da mass shift per hexosamine into the glycans. This technique afforded the significant advantage that all sample preparation procedures after cell culture were performed in the same sample vial, reducing sample preparation variability between normal and heavy samples. However it can only be applied in the living organism, which is quite limited to cell culture.
Endoglycosidase incorporation of 18O into the N-glycan reducing end in 18O-water was recently proposed by Yang et al.34 for relative quantification of N-glycans released from glycoproteins. Enzymatic 18O labelling featured its simplicity of labelling process occurring during enzymatic digestion and nonexistent chromatographic isotope effects.35 Whereas, a mass shift of 2 Da created overlapping isotopic distributions and complicated the peak identification, limiting its further application in quantifying N-glycans in higher mass range.
Hydrazino-s-triazine labelling, a non-reductively derivatization strategy developed in our group for glycan MS analysis,36,37 remarkably enhanced ionization efficiency of N-glycans in electrospray ionization (ESI)-MS. Herein, a MS-based relative quantification strategy was further established by incorporating deuterium stable isotope into this hydrazino-s-triazine labelling into N-glycan. In the structure of deuterated hydrazino-s-triazine, deuterium atoms were all connected to carbon atoms with sp3 hybridization, thus acted out extremely weak acidity. It was difficult to react for the hydrogen or deuterium atoms with such properties under the labelling and separation conditions performed in this work. The design of our derivatization reagents was perfect for avoiding H/D exchange reaction, which as a result, improved the isotopic stability of reagents greatly. Besides the reliability and accuracy of quantification guaranteed by the high purity of derivatization reagents, an obvious increased mass shift as many as 20 Da provided by introducing twenty deuterium atoms greatly simplified glycan identification and the MS data processing. Maltoheptaose (DP7), as well as N-glycans released from ovalbumin were used to prove the feasibility and reliability of this method. Human serum was finally successfully profiled as a representative complex biosample.
2. Experimental section
2.1 Chemicals and materials
DP7 was purchased from TCI America (Portland, OR). Acetic acid (99.9985%) was purchased from Alfa Aesar (Ward Hill, MA). Ammonium formate and ovalbumin were purchased from Sigma Aldrich (St. Louis, MO). Peptide-N-glycosidase F (500 U μL−1), 10× glycoprotein denaturing buffer [5% SDS, 0.4 M dl-dithiothreitol (DTT)], 10% Nonidet P-40 (NP-40) and 10× G7 reaction buffer [0.5 M sodium phosphate (pH 7.5)] were all purchased from New England BioLabs (Ipswich, MA). LC-MS grade water, methanol and acetonitrile (ACN) were obtained from J. T. Baker (Center Valley, PA). Water for other uses was purified by Milli-Q pure water system (Bedford, MA). C18 Sep-Pak cartridges were purchased from Waters (Milford, MA, USA); porous graphitized carbon (PGC) columns were from Grace (Columbia, MD, USA). Human serum from healthy people was provided by the Hospital of Peking University and performed in compliance with the relevant laws and institutional guidelines, and the Hospital of Peking University had approved all the experiments.2.2 Synthesis of labelling reagents
The materials and syntheses of two labelling reagents normal tag 2-hydrazino-4,6-bis-(diethylamino)-s-triazine (HDEAT) and heavy tag 2-hydrazino-4,6-bis-(d10-diethylamino)-s-triazine (d20-HDEAT) were detailed in ESI.†2.3 N-Glycan releasing
10 μL ovalbumin (10 mg mL−1 in water) or human serum was spiked with 20 μL DP7 (1 μM in water) and diluted with 60 μL water, and denatured by adding 10 μL 10× glycoprotein denaturing buffer and heated at 99 °C for 10 minutes. Then the solution was cooled to room temperature and followed by the addition of 20 μL 10% NP-40, 20 μL 10× G7 reaction buffer, 59 μL water and 1 μL PNGase F (500 U μL−1). The reaction mixture was incubated at 37 °C for 18 hours. The enzymatically released N-glycans were purified by reversed-phase solid phase extraction (SPE) and extracted using PGC column. The glycan solution was dried at room temperature in vacuum with concentrator plus (Eppendorf, NY, USA) and stored at −20 °C for further use.2.4 Glycan derivatization
Glycans derivatization using normal and corresponding isotopic labelling reagents were both carried out according to a previously reported method with little modification.32 Specifically, 20 μL normal or heavy hydrazino-s-triazine [50 μM in MeOH/acetic acid (75/25, v/v)] and DP7 (1 pmol) or N-glycans from glycoprotein reacted at 56 °C for 3 hours. The reaction products were then dried under vacuum and re-dissolved in 10 μL HILIC initial condition solution (75% ACN, 25% 10 mM ammonium acetate in water, pH = 4.7). The isotopic and normal labelled N-glycans released from ovalbumin were mixed at different ratios of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 5:1, 2:1, 1:1, 1:2, 1:5 and 1:10 for the following ESI-MS analysis. The labelled N-glycans of human serum were mixed at a ratio of 1:1 for LC-MS analysis without further purification.
:1, 5:1, 2:1, 1:1, 1:2, 1:5 and 1:10 for the following ESI-MS analysis. The labelled N-glycans of human serum were mixed at a ratio of 1:1 for LC-MS analysis without further purification.
2.5 ESI-MS analysis
ESI-MS analysis was performed on Thermo Q-Exactive mass spectrometer equipped with heated electrospray ionization (HESI) source (Thermo Fisher, MA, USA). MS detection range was set as 800 to 2100. The derivative glycans were injected at a flow rate of 5 μL min−1 with a syringe pump. The electrospray was under 3200 V with a stainless capillary heating to 320 °C. In the Orbitrap, the AGC target was set as 1 × 106 ions with a maximum injection time of 50 ms. For the HESI source, the sheath gas was set as 4 arb. units, the auxiliary gas and the heater of auxiliary gas were disabled, and the RF level was set as 50 arb. units. The mass spectrometer was calibrated according to manufacturer protocol and Xcalibur (ver. 2.2) was used for data analysis.2.6 LC-MS analysis
LC-MS analysis was performed on Dionex Ultimate 3000 UHPLC (Thermo Fisher, MA, USA) and Thermo Q-Exactive mass spectrometer. Separation of derivative glycans was performed on a HILIC column, i.e. Waters ACQUITY UPLC BEH Amide column (1.7 μm particle size, 2.1 mm × 75 mm). Mobile phase A and B were 10 mM ammonium formate in water (pH 4.7) and 100% ACN, respectively, and pumped at a constant flow rate of 0.2 mL min−1. 1 μL sample was injected at the initial gradient condition (75% mobile phase B). And the gradient was eluted from 75% to 50% solvent B over 25 min with a total run time of 37 min. For the HESI source, the sheath gas was set as 35 arb. units, the auxiliary gas was set as 10 arb. units, the temperature of auxiliary gas was heated to 310 °C, and the RF level was set as 55 arb. units. The other MS parameters were set as described above.373. Results and discussion
3.1 High isotopic purity of d20-HDEAT
Our self-designed HDEAT and its corresponding deuterated labelling reagent d20-HDEAT were both prepared under quite mild conditions without air-free techniques in the similar synthetic protocol as described in ESI.† The d11-diethylamine replaced diethylamine to participate in the synthesis of d20-HDEAT and introduce a 20 Da mass difference between normal and heavy labelling reagents. The 1H NMR and 13C NMR characterization results of the reagents were presented in ESI (Fig. S1 and S2†). Furthermore, high resolution MS (HRMS) was implemented to calculate isotopic purity of d20-HDEAT. As shown in Fig. 1, the purity of d20-derivative was high to 91.6% accompanying with 8.2% of d19-derivative and only 0.2% d0-derivative reagent.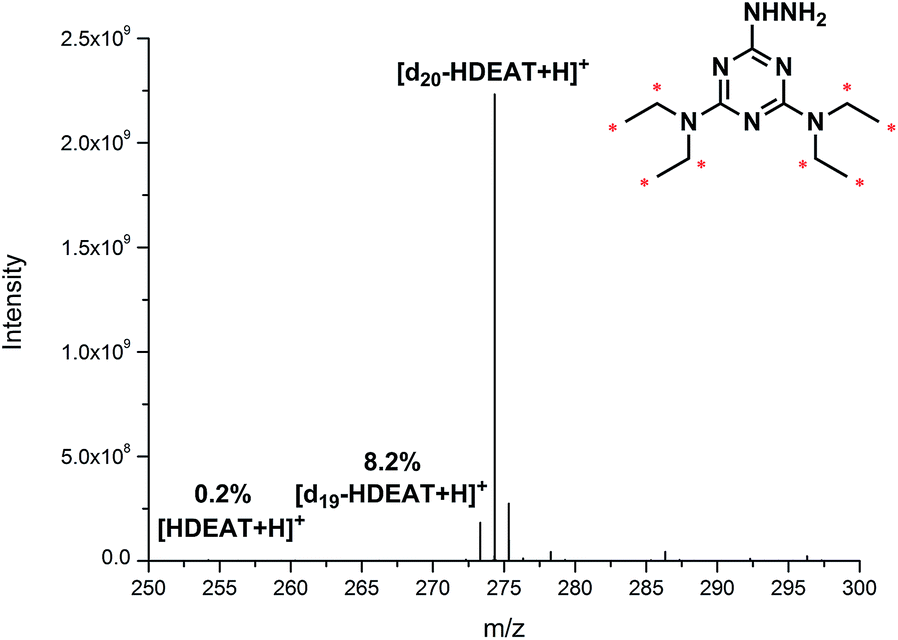 | ||
| Fig. 1 High resolution mass spectrum of d20-HDEAT by the direct infusion. | ||
The high deuterated ratio to 99.6% of commercially available d11-diethylamine facilitated an isotopic purity as high as 90% above of d20-HDEAT, which was nearly impossible for 13C20 or 15N20 derivative reagents with relatively low isotopic purity of 13C or 15N isotopic reagents. Compared with 13C or 15N isotopic labelling reagent, deuterated reagents presented significant advantages with higher isotopic purity and effectively reduced behaviour variability between normal and heavy samples in mass spectra. Moreover, high deuterium purity of isotopic labelling reagent d20-HDEAT also played a crucial role in simplifying mass spectra of labelled glycans and guaranteeing the high accuracy of isotope relative quantification.
3.2 Remarkably simplified glycan identification and data analysis using d20-HDEAT
An inevitable problem existing in glycan isotope relative quantification strategies was isotopic distribution overlapping, especially for high-molecular-weight glycans. Generally, the already fully-developed labelling methods provided mass shift ranging from 1 to 10 Da, which were possible to result in serious overlapping in mass spectra and complicate the data analysis.Herein, we successfully achieved a mass shift as many as 20 Da between heavy tag (d20-HDEAT) and normal tag (HDEAT) and avoided nearly all the possible isotopic distribution overlapping. As shown in Fig. 2, even for high-molecular-weight glycan as large as 10000 Da, isotopic distribution overlapping was still not observed in theoretical simulation mass spectra. Therefore the precision of glycan qualitative identification was greatly enhanced and glycan identification and the data processing were significantly simplified as well. With a remarkable 20 Da mass shift, d20-HDEAT could be widely applied in glycomics especially high accurate glycan isotope relative quantification as a promising deuterated labelling reagent.
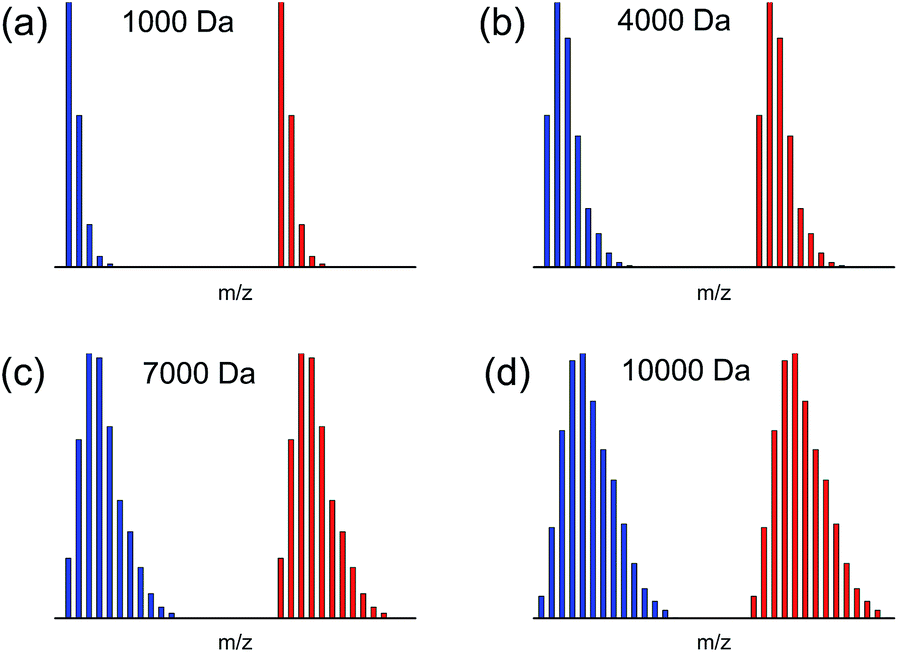 | ||
| Fig. 2 Theoretical simulation of isotopic distributions for HDEAT and d20-HDEAT labelled glycans in 1:1 ratio at (a) ∼1000 Da (Hex7), (b) ∼4000 Da (Hex25), (c) ∼7000 Da (Hex44), (d) ∼10000 Da (Hex62). | ||
3.3 Method validation of the isotope relative quantification strategy
For glycan relative quantification, it is necessary to guarantee that normal and heavy tagged glycans perform the same behaviour with same ionization efficiency in mass spectra, on which condition the data are credible and meaningful. We took maltoheptaose (DP7) as a model standard glycan for method validation to ensure reliability. HDEAT and d20-HDEAT labelled DP7 were mixed at a ratio of 1:1 for ESI-MS and MS2 analysis. The concentration of DP7 labelled by normal and heavy reagents for method validation was 500 nM. We chose such a high concentration because it was important to get MS2 spectra with acceptable intensity. Only by increasing the absolute concentration of parent ions, could the signal intensity of fragments be increased. As shown in Fig. 3, the signal of corresponding fragments tagged with HDEAT and d20-HDEAT in mass spectra remained almost the same, which guaranteed the reliability of relative quantitation. Indirectly, the above results indicated that HDEAT and d20-HDEAT labelled glycans were not only ionized but also fragmented identically. “Y” fragments were observed with consecutive loss of hexose whose molecular weight was 162 Da accompanying with slight dehydration (18 Da loss). These fragments could be clearly identified by a 20 Da mass difference.
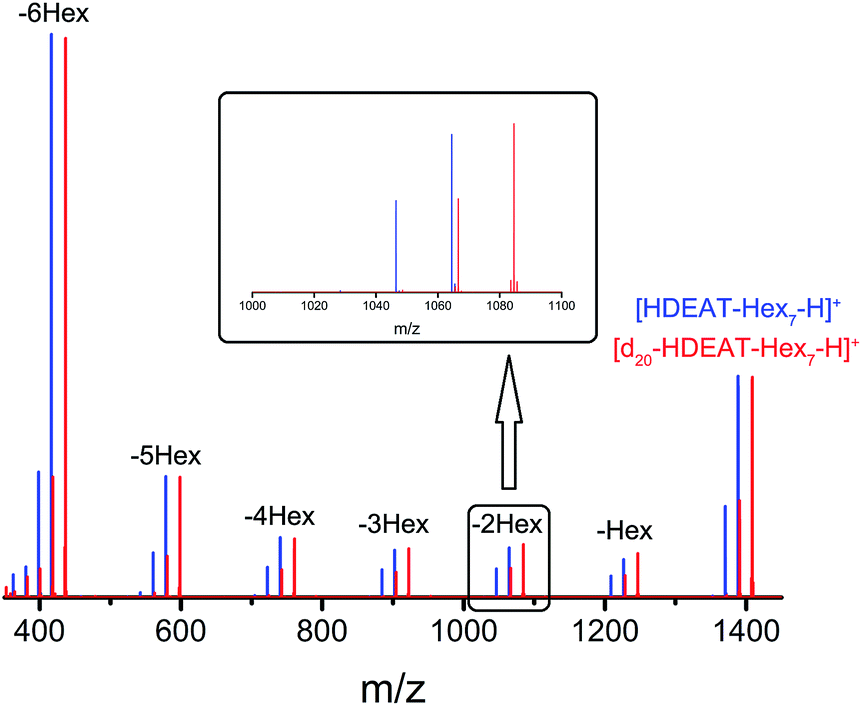 | ||
| Fig. 3 MS2 characterization of normal (HDEAT) and heavy (d20-HDEAT) tags labelled DP7 mixed at a ratio of 1:1. (The inner figure was the enlargement of the fragment of normal and heavy tags labelled DP7 with a loss of two hexose). | ||
To verify the feasibility of isotope relative quantitative analysis, five typical N-glycans (Hex4HexNAc2, Hex3HexNAc3, Hex5HexNAc2, Hex4HexNAc3 and Hex6HexNAc2) enzymatically cleaved from ovalbumin were selected as model analytes. N-glycans labelled with HDEAT and d20-HDEAT were mixed at different ratios (10:1, 5:1, 2:1, 1:1, 1:2, 1:5 and 1:10) for ESI-MS analysis. The dependence of signal ratios (y) of normal and heavy tagged glycans versus the corresponding concentration ratios (x) as shown in Fig. S3 in ESI† was fitted linearly and corresponding calibration equations and correlation coefficients were presented in Table 1. It is obvious to find that the slopes of all glycans were all around 1, which represented the reliability of the isotope relative quantification method. Together with other data in the Table 1, these results demonstrated that the developed method provided a good linear response and 2 orders of magnitude or higher in dynamic range for relative glycan quantitation. Considering the high purity of deuterated labelling reagent, mild derivatization conditions, and satisfactory accuracy, our proposed glycans isotope relative quantification method has proved to be a powerful tool for glycans analysis.
| Glycoform | Calibration equation | R2 |
|---|---|---|
| Hex4HexNAc2 | y = 1.23x − 0.16 | 0.999 |
| Hex3HexNAc3 | y = 1.07x − 0.04 | 0.999 |
| Hex5HexNAc2 | y = 1.13x − 0.07 | 0.999 |
| Hex4HexNAc3 | y = 1.15x − 0.08 | 0.999 |
| Hex6HexNAc2 | y = 1.15x − 0.06 | 0.999 |
3.4 Relative quantification of glycans from complex bio-sample
As for glycan analysis, matrix effects hinder the accurate and reliable quantification of glycans in complex bio-samples. We adopted human serum as complex sample and successfully avoided serious matrix effects by LC separation. DP7 was spiked in human serum as internal standard.32 N-glycans enzymatically cleaved from the same human serum were labelled by HDEAT and d20-HDEAT respectively and mixed at a ratio of 1:1. LC separation was applied to remove the matrix effect before MS analysis. High mannose, complex, fucosylated and sialylated glycans were chosen as representatives of different types of N-glycans. As shown in Fig. 4, isotopic distribution overlapping was not observed both in single and double charged N-glycans as mentioned above. Moreover, corrected by internal standard DP7, the MS response ratios of HDEAT and d20-HDEAT labelled glycans were approximately 1:1 as expected. A total of 48 N-glycans were detected in only 16 minutes with 20 Da mass shifts between HDEAT and d20-HDEAT labelled glycans. A slight retention time shift (<10 s) was observed in HILIC column between HDEAT and d20-HDEAT labelled glycans, which suggested similar ionization condition of the same glycans, as presented in Fig. 5(a). However, a few retention time shift indeed caused problems in separation results. The quantification method based on the extracted ion chromatograms was a good way to solve the inevitable contradiction between LC separation and ionization consistency.38,39 The ion chromatograms of HDEAT and d20-HDEAT labelled N-glycans were extracted respectively and quantification analysis was managed based on the well-shaped extracted ionization chromatograms. As shown in Fig. 5(b), the peaks in blue shadows were the first twenty HDEAT labelled glycans while peaks in red shadows were the first twenty deuterated reagent labelled glycans according to retention time. Most glycans could be extracted clearly and quantified. And the almost equal intensity for both normal and heavy tags labelled samples demonstrated that the few retention time shift would not cause problems for informatics. The N-glycans monitored in human serum were listed in Table S1 in ESI† including their corresponding peak areas and glycan abundance ratio. Table S2 (ESI†) listed the exact mass of the typical glycans presented in the research and their theoretical m/z of corresponding derivatives. The simplification of labelling method and labour-saving data processing made our designed glycans isotope relative quantification method appealing to glycans analysis for complex bio-sample.
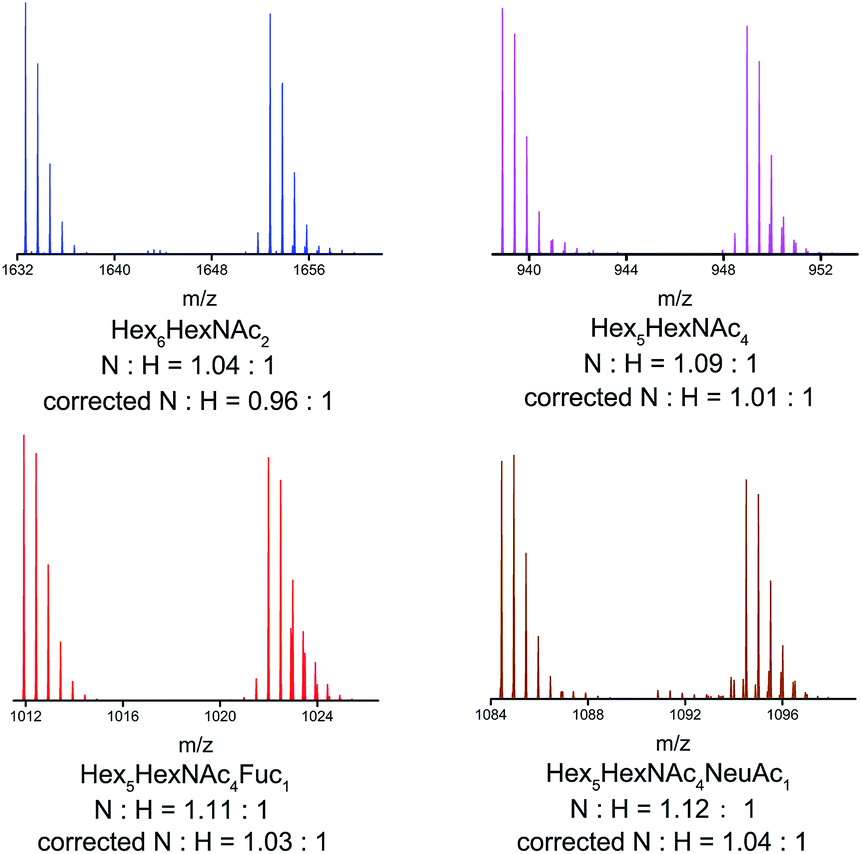 | ||
| Fig. 4 Mass spectra of four N-glycan forms from human serum. ‘N’ was referred to normal tag (HDEAT), and ‘H’ to heavy tag (d20-HDEAT). The ‘N:H’ was the ratio of normal tag and heavy tag labelled glycan pairs. The ‘corrected N:H’ was ‘N:H’ ratio corrected by internal standard. (Hex6HexNAc2 was single charged, and the left were double charged). | ||
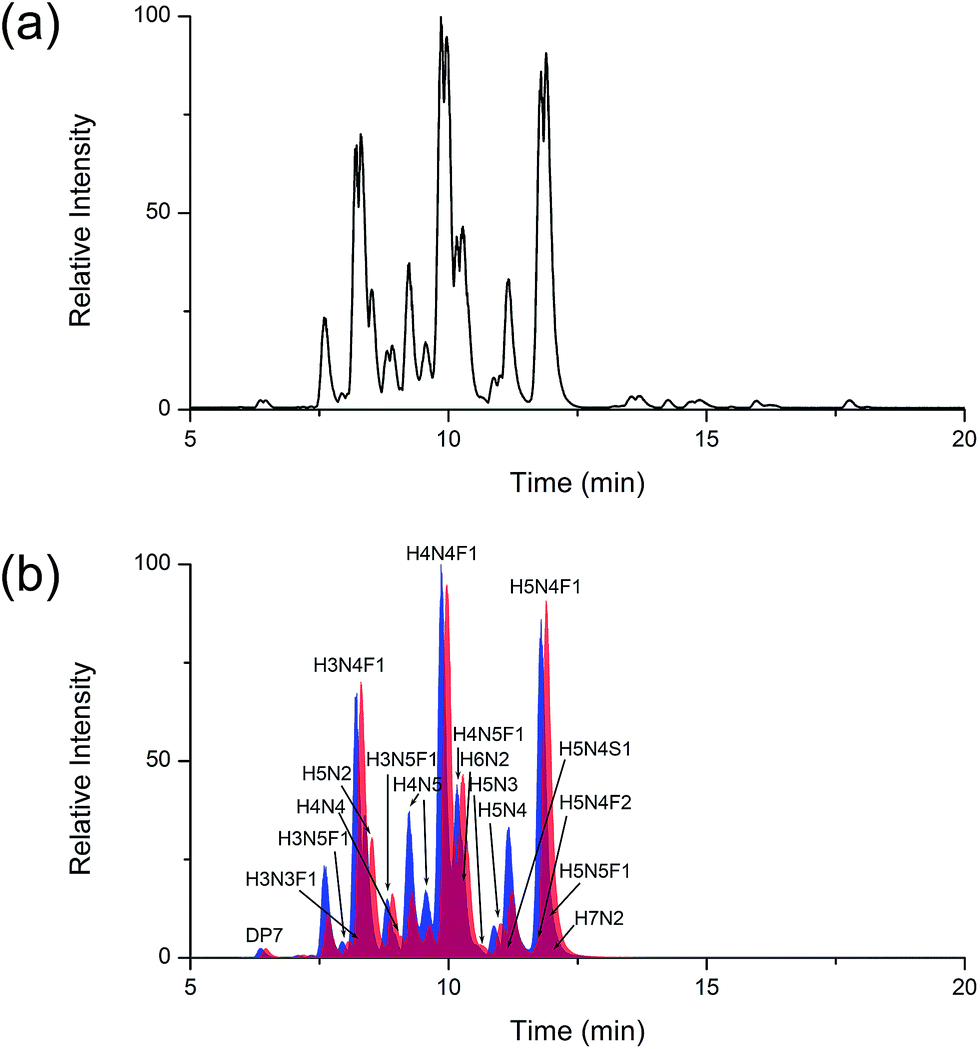 | ||
| Fig. 5 (a) The base peak chromatogram of LC-MS analysis of serum N-glycans and (b) the overlaid first twenty extracted ion chromatograms of HDEAT (blue shadows) and d20-HDEAT labelled N-glycans (red shadows). The ‘H’, ’N’, ‘S’ and ’F’ represented the abbreviations of Hex, HexNac, sialic acid and fucose in peak assignment. | ||
4. Conclusions
Relative quantification via incorporation of stable-isotope labelling is usually necessary for N-glycan quantification by mass spectroscopy, which is always depended on deuterium, 13C, 15N or 18O. The already fully-developed labelling methods always suffered from the low-purity of derivatization reagents or serious overlapping isotopic distributions which complicated the peak identification and data analysis. Our designed deuterated labelling reagent with high isotopic purity, d20-HDEAT, guaranteed the reliability and accuracy of relative quantification. Meanwhile, as many as 20 Da mass shift effectively avoided isotopic distribution overlapping and greatly simplified glycan identification and data analysis especially for high-molecular-weight glycans. Method validation using N-glycans from ovalbumin and human serum confirmed the reliability and feasibility of glycans isotope relative quantification. The successful relative quantification of N-linked glycans had a great potential as a powerful tool to discover glycan biomarkers of N-glycan associated disease diagnosis.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21275009).Notes and references
- R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS PubMed.
- A. Kobata, Glycoconjugate J., 2000, 17, 443–464 CrossRef CAS.
- A. Helenius and M. Aebi, Annu. Rev. Biochem., 2004, 73, 1019–1049 CrossRef CAS PubMed.
- R. B. Parekh, R. A. Dwek, B. J. Sutton, D. L. Fernandes, A. Leung, D. Stanworth, T. W. Rademacher, T. Mizuochi, T. Taniguchi, K. Matsuta, F. Takeuchi, Y. Nagano, T. Miyamoto and A. Kobata, Nature, 1985, 316, 452–457 CrossRef CAS PubMed.
- R. Parekh, I. Roitt, D. Isenberg, R. Dwek and T. Rademacher, J. Exp. Med., 1988, 167, 1731–1736 CrossRef CAS.
- T. Nguyen-Khuong, A. V. Everest-Dass, L. Kautto, Z. Zhao, M. D. P. Willcox and N. H. Packer, Glycobiology, 2015, 25, 269–283 CrossRef PubMed.
- J. Wang, D. Yang, C. Li, S. Shang and J. Xiang, J. Periodontal Res., 2014, 49, 197–204 CrossRef CAS PubMed.
- B. Adamczyk, T. Tharmalingam and P. M. Rudd, Biochim. Biophys. Acta, Gen. Subj., 2012, 1820, 1347–1353 CrossRef CAS PubMed.
- M. M. Fuster and J. D. Esko, Nat. Rev. Cancer, 2005, 5, 526–542 CrossRef CAS PubMed.
- J. Zaia, Mass Spectrom. Rev., 2004, 23, 161–227 CrossRef CAS PubMed.
- J. Zaia, Chem. Biol., 2008, 15, 881–892 CrossRef CAS PubMed.
- H. J. An and C. B. Lebrilla, Mass Spectrom. Rev., 2011, 30, 560–578 CrossRef CAS PubMed.
- I. Ciucanu and F. Kerek, Carbohydr. Res., 1984, 131, 209–217 CrossRef CAS.
- Y. Mechref, P. Kang and M. V. Novotny, in Glycomics: Methods and Protocols, ed. N. H. Packer and N. G. Karlsson, 2009, vol. 534, pp. 53–64 Search PubMed.
- D. J. Harvey, Analyst, 2000, 125, 609–617 RSC.
- A. Guttman, Nature, 1996, 380, 461–462 CrossRef CAS PubMed.
- J. You, X. Sheng, C. Ding, Z. Sun, Y. Suo, H. Wang and Y. Li, Anal. Chim. Acta, 2008, 609, 66–75 CrossRef CAS PubMed.
- E. Lattova and H. Perreault, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 793, 167–179 CrossRef CAS.
- G.-C. Gil, Y.-G. Kim and B.-G. Kim, Anal. Biochem., 2008, 379, 45–59 CrossRef CAS PubMed.
- Y. Zhou, Y. Shan, L. Zhang and Y. Zhang, J. Chromatogr. A, 2014, 1365, 1–11 CrossRef CAS PubMed.
- M. J. Bowman and J. Zaia, Anal. Chem., 2007, 79, 5777–5784 CrossRef CAS PubMed.
- J. Yuan, N. Hashii, N. Kawasaki, S. Itoh, T. Kawanishi and T. Hayakawa, J. Chromatogr. A, 2005, 1067, 145–152 CrossRef CAS PubMed.
- P. Kang, Y. Mechref, Z. Kyselova, J. A. Goetz and M. V. Novotny, Anal. Chem., 2007, 79, 6064–6073 CrossRef CAS PubMed.
- J. A. Atwood, L. Cheng, G. Alvarez-Manilla, N. L. Warren, W. S. York and R. Orlando, J. Proteome Res., 2008, 7, 367–374 CrossRef CAS PubMed.
- M. J. Bowman and J. Zaia, Anal. Chem., 2010, 82, 3023–3031 CrossRef CAS PubMed.
- P. Zhang, Y. Zhang, X. Xue, C. Wang, Z. Wang and L. Huang, Anal. Biochem., 2011, 418, 1–9 CrossRef CAS PubMed.
- C. N. Filer, J. Labelled Compd. Radiopharm., 1999, 42, 169–197 CrossRef CAS.
- B. Xia, C. L. Feasley, G. P. Sachdev, D. F. Smith and R. D. Cummings, Anal. Biochem., 2009, 387, 162–170 CrossRef CAS PubMed.
- C. Michael and A. M. Rizzi, J. Chromatogr. A, 2015, 1383, 88–95 CrossRef CAS PubMed.
- E. Gimenez, V. Sanz-Nebot and A. Rizzi, Anal. Bioanal. Chem., 2013, 405, 7307–7319 CrossRef CAS PubMed.
- J. M. Prien, B. D. Prater, Q. Qin and S. L. Cockrill, Anal. Chem., 2010, 82, 1498–1508 CrossRef CAS PubMed.
- S. H. Walker, J. Budhathoki-Uprety, B. M. Novak and D. C. Muddiman, Anal. Chem., 2011, 83, 6738–6745 CrossRef CAS PubMed.
- R. Orlando, J.-M. Lim, J. A. Atwood III, P. M. Angel, M. Fang, K. Aoki, G. Alvarez-Manilla, K. W. Moremen, W. S. York, M. Tiemeyer, M. Pierce, S. Dalton and L. Wells, J. Proteome Res., 2009, 8, 3816–3823 CrossRef CAS PubMed.
- W. Zhang, H. Wang, H. Tang and P. Yang, Anal. Chem., 2011, 83, 4975–4981 CrossRef CAS PubMed.
- C. Fenselau and X. Yao, J. Proteome Res., 2009, 8, 2140–2143 CrossRef CAS PubMed.
- C. Tie and X.-X. Zhang, Anal. Methods, 2012, 4, 357–359 RSC.
- M.-Z. Zhao, Y.-W. Zhang, F. Yuan, Y. Deng, J.-X. Liu, Y.-L. Zhou and X.-X. Zhang, Talanta, 2015, 144, 992–997 CrossRef PubMed.
- C. C. Nwosu, J. Huang, D. L. Aldredge, J. S. Strum, S. Hua, R. R. Seipert and C. B. Lebrilla, Anal. Chem., 2013, 85, 956–963 CrossRef CAS PubMed.
- J. Huang, H. Lee, A. M. Zivkovic, J. T. Smilowitz, N. Rivera, J. B. German and C. B. Lebrilla, J. Proteome Res., 2014, 13, 681–691 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, figures and tables. See DOI: 10.1039/c5ra12005e |
| This journal is © The Royal Society of Chemistry 2015 |