
An integrated metabonomics and microbiology analysis of host-microbiota metabolic interactions in rats with Coptis chinensis-induced diarrhea†
Yemeng Li‡
a,
Qiongfeng Liao‡b,
Manna Linab,
Danmin Zhonga,
Lin Weia,
Bo Hanc,
Hui Miaoa,
Meicun Yaoa and
Zhiyong Xie*a
aSchool of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, 510006, P. R. China. E-mail: xiezy2074@yahoo.com; Fax: +86 20 39943000; Tel: +86 20 39943047
bSchool of Chinese Materia Medica, Guangzhou University of Chinese Medicine, Guangzhou, 510006, P. R. China
cSchool of Pharmacy, Shihezi University, Shihezi, 832000, P. R. China
First published on 14th September 2015
Abstract
Coptis chinensis Franch., a bererine-containing traditional Chinese medicine (TCM), is often used to treat intestinal infections, diabetes and hyperlipidaemia, and often causes diarrhea. To clarify the potential mechanism of toxicity that induces diarrhea, Sprague-Dawley (SD) rats were treated with Coptis chinensis dosage of 5 g kg−1 for 14 consecutive days. PCR-denaturing gradient gel electrophoresis (PCR-DGGE) was used to monitor the dynamic changes in the gut microbiota, while 1H NMR profiles were applied to reveal the metabolism of host and microflora. In the Coptis chinensis-treated group, decreased short chain fatty acids (SCFAs) and branched chain fatty acids (BCFAs) and increased branched chain amino acids (BCAAs) levels were detected in faeces, whereas increased BCFAs were present in the urine. This finding implied that Coptis chinensis triggered malabsorption and suppressed bacterial fermentation as well as protein degradation. Meanwhile, decreased levels of Bacteroides and Prevotella and elevated levels of Enterobacter and Veillonella in the treatment group were significantly correlated to the urinary and faecal metabolites. Using metabolite-set enrichment analysis (MSEA) and the correlation analysis between significant bacteria and metabolites, the results demonstrated that Coptis chinensis intervention suppressed glycine and serine metabolism which affected the growth of intestinal bacteria. Moreover, the perturbed microbiome consequently influenced the homeostasis of monosaccharides, amino acids, and choline, and energy metabolism of gut microbiota and host. These findings help to elucidate Coptis chinensis intervention and toxicity; simultaneously, this integrated strategy may provide an effective method for the systematic assessment of host responses to TCM or any other botanical-based nutraceuticals.
Introduction
Coptis chinensis Franch. (Huanglian in Chinese, CF), which contains rich alkaloids such as berberine, is a common herb that has been used in traditional Chinese medicines (TCMs) for millennia to treat intestinal infections, particularly bacterial diarrhea.1 In addition, its cholesterol-lowering and hypoglycemic activities2–4 against diabetes and obesity have attracted significant interest in the medical field. Nevertheless, there are only a few reports on the mechanisms of its adverse effects, including diarrhea, cardiac damage,5 and jaundice,3 resulting from higher doses or long-term treatment with CF. In Chinese clinical reports, diarrhea is one of the most common side effects, with a 19% prevalence rate after CF treatment for diabetes.6 Diarrhea often occurs in inflammatory and infectious conditions.7 Inflammatory diarrhea, which is accompanied by abnormalities in ion transport and mucus secretion, can be found in inflammatory bowel disease (IBD); while infectious diarrhea usually results from exposure to pathogenic bacteria (Escherichia coli, Salmonella, Clostridium difficile, etc.) in a tainted environment or excessive antibiotic use. The types of diarrheas mentioned above are thought to have a close relationship with the maladjusted microflora.7,8 Interestingly, our previous study showed that CF-induced diarrhea may relate to the altered gut microbiota via metabonomics analysis of serum and urine.5In recent years, botanical-based nutraceuticals have been used as complementary interventions and have been found to improve metabolic syndromes, such as obesity, diabetes or fatty liver disease,9–11 by regulating maladjusted microbial community. Liquid chromatography-mass spectrometry (LC-MS), gas chromatography-mass spectrometry (GC-MS) and high-resolution NMR spectroscopy (1H NMR) analyses were widely employed to investigate metabolites in various biological matrixes, including exploring the endogenous mechanism by which the microflora is regulated after berberine treatment in HFD-fed rats.2 Although the rats exhibited altered amino acids, fatty acids, glutamine and glutamate metabolic pathways, as revealed by urinary and liver metabolic profiles, there was no definitive result that explained which intestinal bacteria contribute to these changes in host's metabolic pathways. Hence, GC-MS combined with pyrosequencing or real-time PCR along with monitoring the blood glucose and lipid levels were applied to interpret the functions of significant bacteria in berberine-treated group. CF and berberine significantly reduced the proportions of faecal Firmicutes and Bacteroidetes to the total bacteria in high fat diet (HFD)-fed mice.12 Additionally, Blautia and Allobaculum, the putative SCFA-producing bacteria, were observed and accompanied with elevated faecal SCFA concentrations.4 Based on previous studies, we hypothesize that CF-induced diarrhea may be associated with these notable bacteria. Nevertheless, our previous study demonstrated that metabolic profiles of rats with diarrhea were different from the reported profiles. Unfortunately, there is little awareness of whether other bacteria caused this difference and the other side effects of CF that are present when the maladjusted gut microbiota interacts with host.
Metabolite-Set Enrichment Analysis (MSEA13), an extended approach of gene set enrichment analysis (GSEA14), applies univariate and multivariate statistical methods to better interpret complex hypothesis-free metabolic signatures based on MS or NMR spectra of the biological samples. This knowledge-based over-representation approach highlights significant pathways that are influenced by xenobiotics or affected by diseases, and shrinkages numerous potential target metabolites. Thus, the elucidation of the pathophysiology or pharmacodynamics mechanism seems to be clearer and more profound. Therefore, Pontoizeau et al.15 revealed homeostasis and physiological plasticity of several inbred strain rats via MSEA, despite their widespread divergences in metabolites and gut microbiota. Because the gut microbiota closely interacts with ingested food in the intestine via fermentation, putrefaction, hydrolysis, and dehydroxylation, changes in different biological matrixes particularly faeces together with the corresponding alternation in the gut microbiota should provide information that can be used to interpret mammalian–microbial interactions. Firstly, antibiotic-treated (AB) animal is indispensable for the study of dysbacteriosis.16 Swann et al.17 performed fluorescence in situ hybridization (FISH) analysis and 1H NMR spectroscopy in different antibiotic-induced rats that has proven to be feasible methods for ascertaining the influence of the gut microbiota on host's metabolism. In addition, multivariate statistical analyses16 were applied to reveal the correlation between the perturbed gut microbial community and the changes in faecal metabolites following treatment with different antibiotic, which also explained the function of gut microbiota.
Here, we reported a profiling study of urinary and faecal metabolites in SD rats which exposed to an excessive and long-term dose of CF using 1H NMR, and monitored the fluctuations in gut microbiota via 16S rRNA V3 gene PCR-DGGE. In addition, urine and faecal samples were collected at different time points to characterize the dynamic metabolic profiles resulting from CF administration. Finally, metabolic biomarkers derived from metabonomics analyses were associated with significant bacteria to help clarify diarrhea-associated bacteria in CF-treated rats and to improve our understanding of molecular mechanisms underlying host–microbe interactions at multiple levels.
Materials and methods
Rat intervention study and sample collection
Fourteen male Sprague-Dawley (SD) rats (6–8 weeks old, 180–200 g) from the Laboratory Animal Center of Sun Yat-sen University (Guangzhou, P.R. China) were housed at a 12/12 h light-dark cycle with 24 °C and 50–70% humidity. They had free access to water and commercial rodent food unless otherwise specified. After acclimatization for one week, 14 rats were divided equally and randomly into CF-treated and control groups. We implemented animal experiments based on the Institutional Animal Care and Use Committee (IACUC) of Sun Yat-sen University and the Coptis chinensis decoction was prepared according to a previously reported standardized protocol.5 Rats in control group were subjected to gavage with distilled water, while CF-treated rats were orally administered at a dose of 5 g kg−1 weight decoction of Coptis chinensis for 14 consecutive days, respectively. The dosage was determined based on the reference to the Chinese Pharmacopoeia (10 g per human per day, version 2010) and our pre-study results, and at this dosage we observed the main side effect is diarrhea. Urine and faeces samples were collected in Eppendorf tubes on ice from 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :00 to 16:00 on pre-dose day 1 and post-dose day 7, 14, 21. The fresh samples from each group were stored at −80 °C for NMR and microbiological analyses.
:00 to 16:00 on pre-dose day 1 and post-dose day 7, 14, 21. The fresh samples from each group were stored at −80 °C for NMR and microbiological analyses.
Sample preparation for NMR spectroscopy
Urine samples were thawed on ice and 600 μL was mixed with 60 μL of phosphate buffer/D2O (1.5 M Na2HPO4–NaH2PO4, pH 7.4). The buffer contains 0.1% of NaN3 to avoid bacterial contamination and 0.05% TSP to afford a field-frequency lock. The mixtures were centrifuged at 16000g at 4 °C for 10 min to remove any sediment. Faecal extraction used the optimized method described by Wu et al.18 The 600 μL coalescent supernatant was pipetted into 5 mm NMR tubes and 1D 1H NMR spectra were obtained using a Bruker AVIII 600 MHz spectrometer (Bruker Biospin, Germany) at 600.13 MHz and 298 K. The standard pulse sequence (NOESY) with water presaturation for urine and faecal spectrums also referred to prior study.16 2D NMR experiments, including total correlation spectroscopy (TOCSY), J-resolved spectroscopy (JRES) and 1H-13C HSQC, were performed on selected samples to assign the NMR spectra of the metabolites. The metabolites were simultaneously identified based on the Human Metabolome Database (http://www.hmdb.ca/) and metabonomics toolbox (Chenomx NMR Suit 7.6, Chenomx, Canada) as well as published work.19,20
All 1H NMR spectra were manually phase and baseline-corrected and calibrated to TSP at 0.00 ppm using TOPSPIN (V2.1, Bruker Biospin). The spectral region (0.5–9.50 ppm) was segmented into 0.004 ppm chemical shift bins using the AMIX package (V3.9.14, Bruker Biospin). For urine samples, the water signal (4.70–4.95 ppm) and urea signal (5.50–6.25 ppm) were removed prior to analysis. The water signal (4.68–4.95 ppm) in faecal extracts was also discarded to exclude the efficient water suppression. All remaining regions were normalized to the total integrated spectrum before multivariate date analysis.
Molecular biological analysis and data processing
Total bacterial DNA was extracted from faecal samples using a TIANamp Stool DNA Kit (Tiangen, Beijing, P.R. China) in accordance with the manufacturers' instructions with slight modification and then stored at −20 °C for further analysis. Universal primers 357f_GC clamp and 518r targeted the hypervariable V3 region of the 16S rRNA gene and were used to conduct PCR amplification. Initial denaturation was at 95 °C for 3 min, and then a total 25 cycles including denaturation at 95 °C for 1 min, annealing at 55 °C for 1 min, extension at 72 °C for 1 min, and a final extension at 72 °C at 8 min. After confirmation using agarose gels electrophoresis, the PCR products were analysed with a 38–58% gradient DGGE under constant voltage of 70 V for 13 h at 60 °C in 1× Tris–acetate–EDTA (TAE) buffer using the DCode universal mutation detection system (Junyi, Beijing, P.R. China). Silver staining was performed to visualise the changing profile in predominant bacterial profiles and this fingerprint was recorded by a digital camera (Canon, Japan). DGGE images were converted to black-and-white using Adobe Photoshop CS4, and digitized by Quantity One software (version 4.6.2). The relative band intensity was exported as a data matrix for normalization prior to pattern recognition analysis. Meanwhile, the auto search option was used to mark individual bands of each sample lane, followed by necessary manual correction.Statistical analysis and metabolite-strain correlation networks
Multivariate data analysis was conducted by SIMCA-P+ (version 12.0 Umetrics, Sweden) and SPSS software (V20.0, Chicago, USA). The unit variance (UV)-scaled NMR data and DGGE data were analysed separately with principal component analysis (PCA) to investigate the intrinsic similarity or dissimilarity as well as possible outliers in each matrix. Following partial least squares (PLS) and orthogonal projection to latent structure-discriminant analysis (OPLS-DA), the model was validated using a 7-fold cross-validation method and 200 permutation tests, as well as further assessment using CV-ANOVA tests, with a significant level of p < 0.05. The color-coded loading plots were carried out in MATLAB (The Mathworks Inc.; Natwick, MA, version 7.1) based on correlation coefficient values to interpret important metabolites that contribute to the class separation. Meanwhile MSEA,21 an approach to assess whether significant metabolites found in known metabolic pathway maps or databases coincide with the metabolic signature at a certain level of the biochemical pathway, was applied to reduce the massive number of insignificant metabolic pathways. Finally, the covariation analysis between 1H NMR and DGGE data were integrated by O2PLS22 modeling and Pearson's correlation coefficient23 calculation. NMR peaks or DGGE bands with Q2 > 0.6 and |r| ≥ 0.755 were selected to exhibit a bio-network with the Gephi 0.8.2 software.24Cloning and sequencing of specific bands
Bands that were closely correlated with metabolites in the two groups were excised and subjected to further identification by sequencing. Rinsed DNA bands were dissolved in 25 μL TE buffer for 16 h at 4 °C, and then amplified with universal V3 primers without GC clamp. The positive clones were purified and verified as described in previous study.25 After sequencing (Sangon Biotech, Shanghai, China), the results were assembled with Chromas software for homology searches in NCBI GenBank databases using the BLAST tool. Based on BLAST results, reference sequences of phylogenetic neighbor species (up to 90% similarity) were included to confirm the allocation of the purified band sequences to the most probable species. The sequences were deposited in GenBank with the following accession numbers: KR 611915–611917, KR 708629 and KR 611919–611922.Results and discussion
Metabolic changes in urine and faeces following the CF intervention
CF-treated rats displayed diarrhea after 6 days of oral administration and recovered at 7 days after intervention. CF-induced rats also exhibited growth suppression compared to control group, similar to our previous study.5 From NMR metabolic profiles, we detected 85 metabolites in urine samples and 58 components in faecal solutions after CF consumption by combining CF-induced group with control group (ESI Fig. S1†). The signals from urine mainly contained glucose, glycogen, amino acids, amines, organic acids, TCA intermediate metabolites such as citrate, 2-oxoglutarate, succinate and fumarate, and a series of SCFAs. Faecal spectra were mainly comprised of amino acids, glucose, hemicellulosic sugars (arabinose and xylose), some keto acids (e.g., α-ketoisovalerate and α-ketoisocaproate), as well as amines and SCFAs. The specific NMR assignment can be found in ESI Tables S1 and S2.†Subsequently, spectra signals were converted into a digital matrix for statistical analysis, which could reveal more detailed information about the CF-induced metabonomic changes. Compared with the analysis of day −1 of control and CF-treated group, we found an obvious discrimination between control rats and CF-treated rats on day 7 using PCA model (ESI Fig. S2A and B†). In addition, OPLS-DA models for urinary profile (R2X = 0.528, Q2 = 0.836) and faecal extract spectra (R2X = 0.786, Q2 = 0.963) on day 7 were used to investigate the differences in metabolic concentrations between samples which obtained from CF-induced rats and the matched controls. The OPLS-DA scores plots and the coefficient loading plots (Fig. 1A and B) illustrated that the CF intervention significantly altered metabolites. As Jiang et al.26,27 reported, organ especially liver concentrations of berberine or its metabolites was 10-fold or 30-fold higher than that in plasma, whereas only 0.0939% and 22.74% recovered rate of berberine in urine and feces after oral administration. It is worth mentioning that the differentiation occurred in control group at day −1 and day 7 (ESI Fig. S2C and D†) which suggested that dietary and gavage cause the overall metabolic alteration in control rats over time. To minimize unpredictable factors, we reviewed those biomarkers found on day 7. In addition to verification with VIP values, Student's t-test was used to analyse authentically different metabolites by comparing control samples at day −1 and day 7. Finally, 21 significantly different metabolites in urine samples were sorted out. Those discarded components which expressed differentially in the control group were supposed to have little association with CF administration. Similarly, after removing hypocritical biomarkers from faecal extract solutions, we noticed that 9 faecal metabolites showed significantly lower NMR response after CF perturbation, whereas 10 metabolites in faeces displayed the opposite behavior as shown in Fig. 1.
 | ||
| Fig. 1 1H NMR coefficient loading profiles from OPLS-DA model on day 7 and PLS-DA scores plots with time-dependent trajectory. (A) is for urine samples, (B) is for faeces and in (C) graph: (1), (2), (3) exhibit fluctuations of 4, 3, 3 time points in urine, faeces and microbiota respectively. | ||
To investigate the time-dependence and recoverability of the CF effects, we analysed the CF-induced urinary and faecal metabolic alterations at four and three time points respectively (Fig. 1C). PLS-DA scores plots ((1) R2X = 0.327, Q2 = 0.402; (2) R2X = 0.621, Q2 = 0.906; (3) R2X = 0.776, Q2 = 0.973) showed the specific profiles in the CF-induced group on the selected days. Once the diarrhea occurred, the metabolic trajectories of host and gut microbiota diverged from their initial metabolic position. After 7 days (on day 21) of recovery, we found their metabolic profiles returned to approximately the pretreatment level. On the other hand, using microbial profiles in faecal samples obtained from DGGE analysis, the CF-induced fluctuations in the composition of gut microbiota were in accord with the faecal metabolic changes (Fig. 1C).
Altered metabolites in urinary and faecal samples are related to CF intake
In Fig. 2, significant metabolites in urinary metabolic profile were investigated following treatment and during convalescence, and compared to predose profiles. Alanine, pyruvate and glucose, which are involved in alanine-glucose cycle,28 displayed increased levels in CF-induced group. Elevated levels of choline and glucose were also found in a number of CF intervention urine samples, which were similar to faecal samples (Fig. 3). Branched chain fatty acids (BCFAs) especially isobutyrate, isovalerate, and 2-methylbutyrate are products from oxidation of valine, leucine, and isoleucine, respectively.29 It is also believed that colonic bacteria are capable of decomposing proteins, peptides, and amino acids to produce BCFAs,30 as well as SCFAs. What surprised us is that there were higher levels of BCFAs particularly isobutyrate and isovalerate in CF-administered rats compared to controls, but these differed from faecal metabolism. The increased BCFAs in urine samples implied that host increased BCFAs absorption following CF stimulation to maintain energy balance. Whereas, decreased BCFAs and increased BCAAs of faecal samples suggested that the BCFA–BCAA (branched chain amino acid) pathway has been disturbed, in that the maladjusted gut microbiota decreased the utilization of BCAA to produce BCFA. It is remarkable that urinary hippurate, as a gut microbial-mammalian co-metabolite that is generated from aromatic compounds and polyphenolics by gut microbes,31 was obviously reduced in rats with diarrhea. The dysbiosis of the gut microflora in treated rats not only related to imbalanced SCFAs in faeces but also reflected the decreased hippurate in the urine. | ||
| Fig. 2 Distributions of intensities for selected urinary metabolites based upon the normalized bucket table. Significant metabolites of rats' urine which compared control group with CF-treated group at different time points: day −1 is pre-administration day, day 7 and day 14 are diarrhea days, and day 21 is post-administration day for 7 days. | ||
 | ||
| Fig. 3 Distributions of intensities for selected faecal metabolites based upon the normalized bucket table. Significant metabolites of faeces samples from CF-treated group were compared with control group ones at different time points: day −1 is pre-administration day, day 7 is diarrhea at the first day. | ||
The fecal metabolic profile (Fig. 3), which is a direct reflection of changes in microbial composition due to the CF intervention, showed altered faecal metabolites with previous VIP value sifting and t test (p < 0.05). Butyrate, the most important component of SCFAs, plays a crucial role as an energy source for colon cells.32 Moreover, butyrate provides ATP, which participates in Na+–H+ exchange to promote absorption of water and sodium,33 and this facilitates foundation of the anti-diarrhea hypothesis. In this study, the significantly reduced butyrate level in CF-treated rats may be closely associated with suppressed butyrate-producing bacteria. Choline metabolites such as dimethylamine and trimethylamine, which often modulate lipid metabolism and glucose homeostasis,34,35 had a similar decreasing trend in the group with diarrhea. In contrast, choline level was not consistent with the dynamic change in its metabolites, and instead showed an increase. We speculated that the CF-induced dysbiosis of gut microbiota impaired choline metabolism. On the other hand, the elevated level of ethanolamine from choline pathway is able to facilitate lecithin synthesis and preserve cellular integrity, because cytomembrane consists of lecithin.36 As we know, α-keto acids derived from deaminisation of α-amino acids can form nonessential amino acids or offer energy for host through complete oxygenolysis; while glucogenic amino acids are able to transform into glucose.28 Interestingly, we observed that α-keto acids such as 3-methyl-2-oxovalerate, α-keto-β-methyl-valerate, α-ketoisocaproate, and α-ketoisovalerate, were depleted levels in CF-treated group, whereas some amino acids were consistent with the elevated level of glucose. Therefore, we tentatively hypothesize that these results act as one factor that promotes the growth suppression of CF-treated rats in the current study. Although some urinary and faecal biomarkers have been found to be influenced by the CF intervention, there was no distinct identification of the systematic changes in host.
MSEA focused on meaningful signaling pathways that regulate metabolism
MSEA13 is often applied to quantitative metabolomic data for identifying and interpreting changes in human or other mammalian pathway-associated metabolite concentrations. According to precious studies,21,37 we aimed to determine which pathways were dramatically affected based on the detected metabolites. Enrichment tests with a qualitative overrepresentation analysis or a quantitative enrichment analysis were performed, and numbers of potential target metabolites from KEGG database (http://www.genome.jp/kegg/) were swift reduced into a set of particular metabolic pathways (Fig. 4 and Tables S3–S6†). From metabolite sets enrichment profiles and pathway impact illustrations, we focused on 10 significant pathways in urine and faecal samples. Remarkably, they shared 5 pathways to some extent, although metabolites involved in these shared pathways were not absolutely uniform.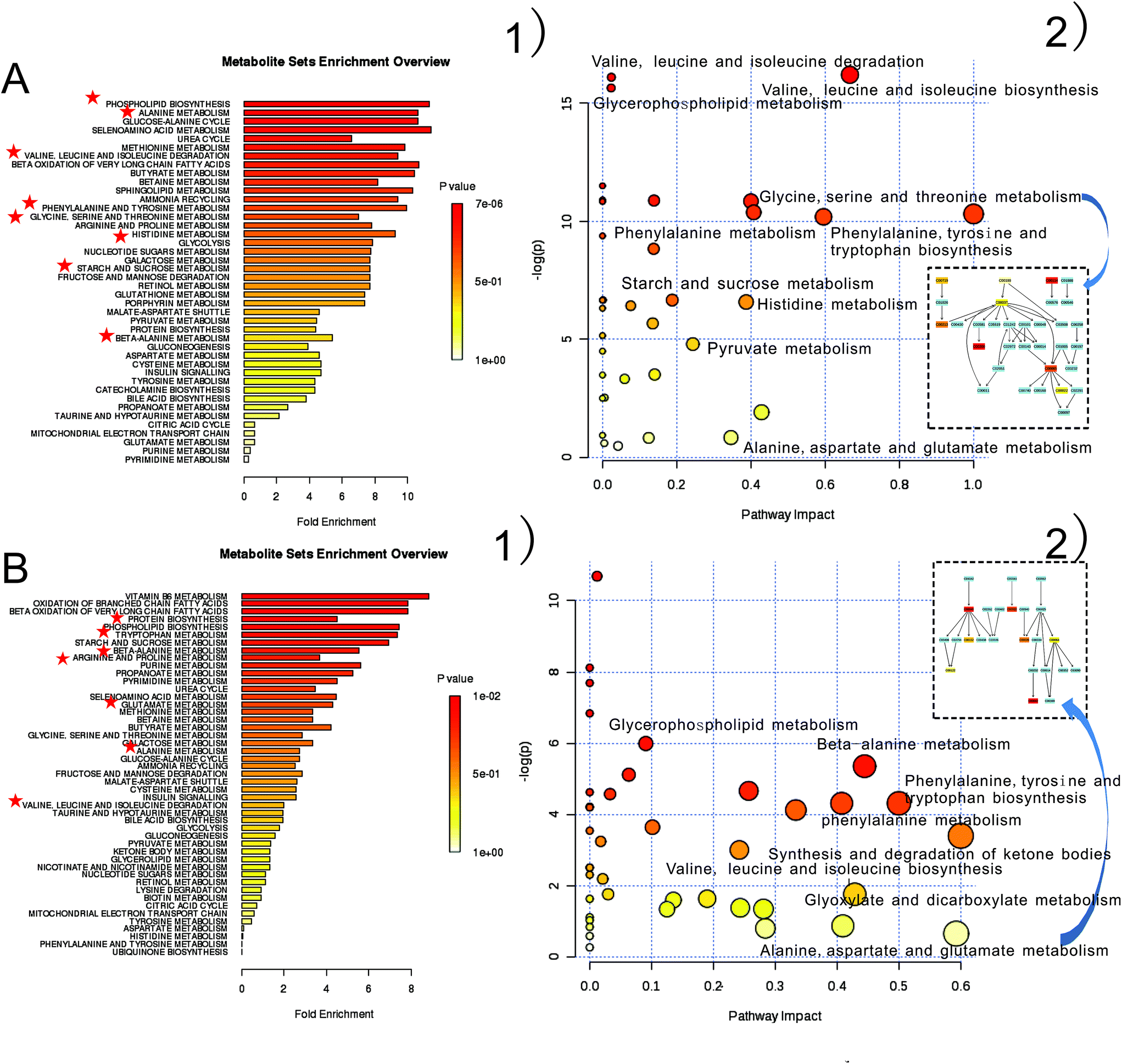 | ||
| Fig. 4 Meaningful metabolic pathways of urine and faeces from MSEA. 57 components in faeces were imported to MSEA to show really changed pathways (A-1), a weight distribution of these pathways displayed on the right hand (A-2). 85 urinary metabolites were conducted as faecal samples, and results exhibited in (B-1 and B-2) severally. At the right corner of each block scheme, it emerged the specific pathway which detected metabolites were involved in (rectangles colored in blue are no metabolites matched, vice versa). | ||
In this study, urinary pathways of host and faecal pathways of intestinal microbiome had an interconnected role in energy metabolism. For instance, pyruvate metabolism, the core of TCA cycle, showed differences in CF-treated faeces. As we know, TCA cycle is the common and ultimate pathway for the oxidation of carbohydrates, fatty acids, and amino acids38 that connects with numerous other pathways. Alanine, aspartate, and glutamate degradation include TCA intermediates such as 2-oxoglutarate, citrate, and succinate showed suppression in urinary samples, as well as glyoxylate and dicarboxylate metabolism. Furthermore, starch and sucrose metabolism was disturbed in view of increased glucose level. These results indicated that host and gut microbiota interacted to maintain balance of energy metabolism via some important metabolic intermediates when suffered from excessive CF. Meanwhile, the maladjusted gut microbiota may relate with some perturbed amino acids pathways. Valine, leucine and isoleucine metabolism were altered in both faeces and urine. Increased leucine and valine levels indicated that their biosynthesis restrained its degradation, and the homologous keto acids such as α-keto-β-methyl-valerate and α-ketoisovalerate, were subsequently decreased. In addition, succinate was affected by the aberrant metabolism because it requires valine and leucine to take part in TCA cycle.39 Excessive and long-term dose of CF not only cut down the energy supplement of host and gut microbiota, it may also tempt invasive bacteria to worsen the systemic metabolism. Aromatic amino acid metabolism includes phenylalanine, tyrosine and tryptophan metabolism, and is required to produce neurotransmitters; it is believed that this gut microbiota-host co-metabolism is associated with the gut-brain axis (GBA).40 The GBA seems to modulate homeostasis of gastrointestinal (GI) tract function once the central nervous system induces changes or the GI tract alters the habitat and perturbs the gut microbiota, because the habitat of the microbiota depends on GI motility and epithelial functions. Significant decrease levels of hippurate and 4-hydroxyhippurate at day 7 and day 14 in the CF-induced group while the elevation at day 21 were observed in urine, which suggested that phenylalanine pathway affected GI tract function and disturbed gut microbiota. However, decreased 2-oxoglutarate level and increased urea and asparagine levels involved in alanine, aspartate and glutamate metabolism demonstrated that excessive CF did not injure the liver and kidney of rats, in that the metabolism correlates with the liver and kidney functions about ammonia transportation.28 Changes in glycine, serine and threonine metabolism (Fig. 4A-2), the additional block scheme) were noticed for glycine, which belongs to the choline degradation pathway and is degraded into creatine through betaine and N,N-dimethylglycine.41 Additionally, glycine and serine can be transformed into one carbon unit which is indispensable for the synthesis of purine and pyridine.28 We found this metabolism was suppressed, which supposed to be the reason that some intestinal bacteria growth was restrained. At the same time, we noticed that phosphorylcholine level in choline degradation pathway increased while dimethylamine and trimethylamine obtained from microbes degradation was dramatically reduced. In short, these aberrant metabolites or pathways were deemed to correlate with altered intestinal bacteria.
Gut microbial markers in CF-induced rats
To investigate CF-induced fluctuations in rats' gut microbiota, we performed DGGE analysis to visualize microbial diversity in faecal samples collected on day 7. R2X and Q2 values from OPLS-DA model (Fig. 5) of digital DGGE gel are 0.564 and 0.907 respectively, which provided a good prediction of discriminating groups. We observed approximately 45 bands, and then integrated 1H NMR spectral data with DGGE microbial data using O2PLS. Thirteen differential bands were associated with significant metabolites (Fig. 6); and only 8 particularly prominent bands were selected for cloning and sequencing (Table 1) (due to segmentary correlations with sporadic metabolites, the 5 residual bands have not been probed in following discussion.). Although the number of bands in CF-treated group has been significantly reduced, there were 4 bacterial species that aroused in rats with diarrhea and 4 bacteria in control group that appeared to correlate with important metabolites. Using BLAST analysis, we obtained bacterial species or phylotypes which were confirmed by clustering analysis, and there were one Firmicute (Veillonella parvula), one Bacteroidete (Bacteroides vulgatus), and two Proteobacteria species (Acidovorax avenae and Enterobacter aerogenes) in CF-treated group, while 4 species from control rats belonged to Bacteroidete. | ||
| Fig. 5 16S rDNA PCR-DGGE on day 7, the red marked bands are significant in diarrhea progressing. OPLS-DA coefficient loading profile (right) displayed the discriminative DGGE bands by comparing the treated group with control. | ||
 | ||
| Fig. 6 Correlation network between altered DGGE bands (the corresponding bacterial species in Table 1) and changed metabolic components of rats' urine and faeces visualized with Gephi. This map calculated using Pearson correlation coefficients (|r| ≥ 0.755 and p < 0.05), and node size denotes the highest effect size for each band or metabolite comparing treated group with control group. Nodes colored red for bands, amaranth for faecal metabolites and cyan for urinary ones. Red lines correspond to positive correlations, whereas blue lines correspond to negative correlations. | ||
| DGGE band | Sample | Seq. length (bp) | Domain | Phylum | Class | Order | Family | Genus | Species | S_ab score | Accession numbers |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a CF, CF-induced group; Ctr, control group. | |||||||||||
| Band 2 | CF | 189 | Bacteria | Bacteroidetes | Bacteroidia | Bacteroidales | Bacteroidaceae | Bacteroides | Bacteroides vulgatus | 1.00 | KR611915 |
| Band 4 | CF | 194 | Bacteria | Proteobacteria | Betaproteobacteria | Burkholderiales | Comamonadaceae | Acidovorax | Acidovorax avenae | 0.90 | KR611916 |
| Band 6 | Ctr | 189 | Bacteria | Bacteroidetes | Bacteroidia | Bacteroidales | Prevotellaceae | Prevotella | Prevotella dentalis | 0.96 | KR611917 |
| Band 10 | CF | 194 | Bacteria | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae | Enterobacter | Enterobacter cloacae complex | 0.96 | KR708629 |
| Band 30 | CF | 195 | Bacteria | Firmicutes | Negativicutes | Selenomonadales | Veillonellaceae | Veillonella | Veillonella parvula | 0.94 | KR611919 |
| Band 36 | Ctr | 190 | Bacteria | Bacteroidetes | Bacteroidia | Bacteroidales | Prevotellaceae | Prevotella | Prevotella denticola | 0.94 | KR611920 |
| Band 39 | Ctr | 189 | Bacteria | Bacteroidetes | Bacteroidia | Bacteroidales | Bacteroidaceae | Bacteroides | Bacteroides helcogenes | 0.94 | KR611921 |
| Band 43 | Ctr | 189 | Bacteria | Bacteroidetes | Bacteroidia | Bacteroidales | Bacteroidaceae | Bacteroides | Bacteroides vulgatus | 0.99 | KR611922 |
CF-induced intestinal bacteria dominated the fluctuation in urinary and faecal metabolites
The maintenance of human health and the occurrences or developments of disease are deemed to have close relationship with gut microbiota. To ascertain the complicated interaction between host and microbes, we integrated 1H NMR spectral data and DGGE microbial data to demonstrate the mechanism of CF-induced diarrhea (Fig. 6). In rats with diarrhea, we found that the dysbiotic signature was highly characteristic for the abundant emergence of Enterobacter aerogenes (band 10) and Veillonella parvula (band 30), which correlated with numbers of faecal metabolites. Enterobacter aerogenes is a normal bacterium and often inhabits in the mammalian intestine, and it is believed to be linked with the occurrence of colicky in infants.42 In the correlation map, Veillonella parvula, a member of the most active microflora that participates in immunoregulation,43 such as Enterobacter, had similar interactions with perturbed metabolites. Two other invasive bacterial species in CF-induced rats were Bacteroides vulgatus (band 2) and Acidovorax avenae (band 4), both of which interfered urine and faeces metabolism. Acidovorax avenae is abundant in plant, but some researchers reported that Comamonadaceae including Acidovorax were decreased in the gut of high weight hosts.44 Interestingly, the suppressed growth of CF-treated rats exactly correlated with increased Acidovorax avenae; once the animals resumed their growth, this bacterium was dramatically reduced (Fig. S3†). Bacteroides, which are obligatory anaerobic bacteria, are important for food digestion because they are rich in carbohydrate transport and protein-metabolism-related enzyme, and contain glycan, vitamins, and cofactor enzymes.45 Bacteria from Bacteroidetes are the predominant and most robust bacteria in the gut of conventional mice, which was ultimately confirmed by our results. Excessive CF impeded these dominant bacteria (band 6, 36, 39 and 43), while the relevant metabolism of CF-induced rats recovered after terminating the intervention.CF-induced diarrhea due to the intrusion of harmful bacteria and the suppression of helpful microbial population, which is analogous to antibiotic treatments, can selectively sterilize normal bacteria. It is different that the dysbiotic microflora in CF-treated rats returned to normal after 7 days of recovery (Fig. S3†), which does not occur in antibiotics treatments.17,63 Further investigations should be performed to access the antimicrobial mechanism of CF because antibiotics often promote the expression of antibiotic resistance genes in the microbiome.70 Antibiotics not only cause dysbacteriosis but often down-regulate the levels of SCFAs, BCFAs, methylamines, xanthine, uracil hippurate etc.40 However, monosaccharides, BCAAs, aromatic amino acids and choline were up-regulated in our CF-induced intestinal turbulence. What amazed us is that the SCFA levels in urine showed an increasing trend which differed from faeces. This distinction also found in antibiotics-treated metabolic profiles,40 because faecal metabolites primarily reflected the gut microbiota metabolism but urinary excretions usually come from the host-microbiome co-metabolism. Metabolic perturbations related to changes in physiological status are only present in urine. Thus, significant components in urine may not be the direct metabolic products of gut microbiota but are altered along with other metabolites in host's systemic circulation.
Conclusion
This work describes toxicity of CF in rats with diarrhea, and the variations in urinary and faecal metabolites have been clarified by an NMR metabolomic study, while the expected dysbiosis of gut microbiota were confirmed by DGGE analysis. Furthermore, we employed covariation analyses of NMR and DGGE data to explore the impact of the altered gut microbial on metabolic profiles of CF-treated rats. The findings indicate that excessive and long-term CF administration perturbed some metabolic pathways such as glycine and serine degradation, and then disturbed the harmonious growth of intestinal bacteria. In rats with diarrhea, Bacteroides and Prevotella were significantly suppressed, while Acidovorax, Enterobacter and Veillonella were promoted. The disrupted gut microflora may have caused the diarrhea in response to the decreased SCFA levels, which play an irreplaceable role in regulating the absorption of water and sodium. Enterobacter and Veillonella have indispensable roles in gut microbiota or host metabolism due to their frequent and compact correlations with the alternations in energy metabolism, amino metabolism and choline metabolism in faeces. Bacteroides and Acidovorax, which are related to most of urinary metabolites such as BCAAs, hippurate, creatine and glutamate, restructure host's metabolic phenotype after CF administration. According to this study, we found that CF toxicity on gut is reversible using metabolic and DGGE trajectory chart. In conclusion, this work inspired us to find a novel way to study drug toxicity. Moreover, global system biology combined with metabonomics can yet be regarded as a commendable strategy for TCM research.Conflict of interest
The authors declare no competing financial interest.Abbreviations
| PCR-DGGE | Polymerase chain reaction-denaturing gradient gel electrophoresis |
| BCFAs | Branched chain fatty acids |
| GC-MS | Gas chromatography-mass spectrometry |
| LC-MS | Liquid chromatography-mass spectrometry |
| NMR | Nuclear magnetic resonance |
| TSP | Sodium 3-(trimethylsilyl) propionate-2,2,3,3-d4 |
| HSQC | 1H-13C Heteronuclear single quantum correlation spectroscopy |
| TCA | Tricarboxylic acid cycle |
| VIP | Variable importance to the projection |
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 81173564, No. 81274028, No. 81473319 and No. 81473540), the National Key Technology R&D Program during the Twelfth Five-Year Plan Period of People's Republic of China (No. 2013BAD10B04-2) and the Guangzhou Science and Technology Program (No. 2014J4100176).References
- J. Tang, Y. Feng, S. Tsao, N. Wang, R. Curtain and Y. Wang, J. Ethnopharmacol., 2009, 126, 5–17 CrossRef CAS PubMed.
- Z. Jiang, F. Liu, E. S. Ong and S. Li, Metabolomics, 2012, 8, 1052–1068 CrossRef CAS.
- R. Shi, H. Zhou, B. Ma, Y. Ma, D. Wu, X. Wang, H. Luo and N. Cheng, Biopharm. Drug Dispos., 2012, 33, 135–145 CrossRef CAS PubMed.
- X. Zhang, Y. Zhao, M. Zhang, X. Pang, J. Xu, C. Kang, M. Li, C. Zhang, Y. Zhang, X. Li, G. Ning and L. Zhao, PLoS One, 2012, 7, e42529 CAS.
- Y. Zhou, Q. Liao, M. Lin, X. Deng, P. Zhang, M. Yao, L. Zhang and Z. Xie, PLoS One, 2014, 9, e88281 Search PubMed.
- X. Zhao, X. Tong, L. Zhao, Q. Zhou, Z. Peng and B. Pang, Zhongguo Zhongyao Zazhi, 2013, 38, 546–547 CAS.
- M. G. Gareau and K. E. Barrett, Curr. Opin. Pharmacol., 2013, 13, 895–899 CrossRef CAS PubMed.
- V. C. Antharam, E. C. Li, A. Ishmael, A. Sharma, V. Mai, K. H. Rand and G. P. Wang, J. Clin. Microbiol., 2013, 51, 2884–2892 CrossRef PubMed.
- J. M. Hollander and J. I. Mechanick, J. Am. Diet. Assoc., 2008, 108, 495–509 CrossRef PubMed.
- G. Xie, A. Zhao, L. Zhao, T. Chen, H. Chen, X. Qi, X. Zheng, Y. Ni, Y. Cheng, K. Lan, C. Yao, M. Qiu and W. Jia, J. Proteome Res., 2012, 11, 3449–3457 CrossRef CAS PubMed.
- T. E. Cowan, M. S. Palmnäs, J. Yang, M. R. Bomhof, K. L. Ardell, R. A. Reimer, H. J. Vogel and J. Shearer, J. Nutr. Biochem., 2014, 25, 489–495 CrossRef CAS PubMed.
- W. Xie, D. Gu, J. Li, K. Cui and Y. Zhang, PLoS One, 2011, 6, e24520 CAS.
- J. Xia and D. S. Wishart, Nucleic Acids Res., 2010, 38(suppl.), W71–W77 CrossRef CAS PubMed.
- A. Subramanian, P. Tamayo, V. K. Mootha, S. Mukherjee, B. L. Ebert, M. A. Gillette, A. Paulovich, S. L. Pomeroy, T. R. Golub, E. S. Lander and J. P. Mesirov, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15545–15550 CrossRef CAS PubMed.
- C. Pontoizeau, J. F. Fearnside, V. Navratil, C. Domange, J. B. Cazier, C. Fernández-Santamaría, P. J. Kaisaki, L. Emsley, P. Toulhoat, M. T. Bihoreau, J. K. Nicholson, D. Gauguier and M. E. Dumas, J. Proteome Res., 2011, 10, 1675–1689 CrossRef CAS PubMed.
- Y. Zhao, J. Wu, J. V. Li, N. Y. Zhou, H. Tang and Y. Wang, J. Proteome Res., 2013, 12, 2987–2999 CrossRef CAS PubMed.
- J. R. Swann, K. M. Tuohy, P. Lindfors, D. T. Brown, G. R. Gibson, I. D. Wilson, J. Sidaway, J. K. Nicholson and E. Holmes, J. Proteome Res., 2011, 10, 3590–3603 CrossRef CAS PubMed.
- J. Wu, Y. An, J. Yao, Y. Wang and H. Tang, Analyst, 2010, 135, 1023–1030 RSC.
- C. A. Merrifield, M. Lewis, S. P. Claus, O. P. Beckonert, M. E. Dumas, S. Duncker, S. Kochhar, S. Rezzi, J. C. Lindon, M. Bailey, E. Holmes and J. K. Nicholson, Mol. BioSyst., 2011, 7, 2577–2588 RSC.
- G. le Gall, S. O. Noor, K. Ridgway, L. Scovell, C. Jamieson, I. T. Johnson, I. J. Colquhoun, E. K. Kemsley and A. Narbad, J. Proteome Res., 2011, 10, 4208–4218 CrossRef CAS PubMed.
- M. E. Dumas, J. Kinross and J. K. Nicholson, Gastroenterology, 2014, 146, 46–62 CrossRef PubMed.
- M. Rantalainen, O. Cloarec, O. Beckonert, I. D. Wilson, D. Jackson, R. Tonge, R. Rowlinson, S. Rayner, J. Nickson, R. W. Wilkinson, J. D. Mills, J. Trygg, J. K. Nicholson and E. Holmes, J. Proteome Res., 2006, 5, 2642–2655 CrossRef CAS PubMed.
- D. J. Crockford, E. Holmes, J. C. Lindon, R. S. Plumb, S. Zirah, S. J. Bruce, P. Rainville, C. L. Stumpf and J. K. Nicholson, Anal. Chem., 2006, 78, 363–371 CrossRef CAS PubMed.
- S. Bastian Heymann and M. Jacomy, International AAAI Conference on Weblogs and Social Media; Association for the Advancement of Artificial Intelligence, CA, Palo Alto, 2009 Search PubMed.
- H. Lu, J. He, Z. Wu, W. Xu, H. Zhang, P. Ye, J. Yang, S. Zhen and L. Li, Microb. Ecol., 2013, 65, 781–791 CrossRef PubMed.
- X. Tan, J. Ma, R. Feng, C. Ma, W. Chen, Y. Sun, J. Fu, M. Huang, C. He, J. Shou, W. He, Y. Wang and J. Jiang, PLoS One, 2013, 8, e77969 CAS.
- J. Ma, R. Feng, X. Tan, C. Ma, J. Shou, J. Fu, M. Huang, C. He, S. Chen, Z. Zhao, W. He, Y. Wang and J. Jiang, J. Pharm. Sci., 2013, 102, 4181–4192 CrossRef CAS PubMed.
- X. Cha and C. Zhou, in Biochemistry, ed. N. Cheng, and A. Zhou, People's Medical Publishing House, Beijing, 2008, pp. 179–206 Search PubMed.
- E. A. Smith and G. T. Macfarlane, Anaerobe, 1997, 3, 327–337 CrossRef CAS PubMed.
- M. Blaut and T. Clavel, J. Nutr., 2007, 137(suppl. 2), 751–755 Search PubMed.
- J. K. Nicholson, E. Holmes and I. D. Wilson, Nat. Rev. Microbiol., 2005, 3, 431–438 CrossRef CAS PubMed.
- S. E. Pryde, S. H. Duncan, G. L. Hold, C. S. Stewart and H. J. Flint, FEMS Microbiol. Lett., 2002, 217, 133–139 CrossRef CAS PubMed.
- J. H. Cumming, J. L. Rombeau and T. Sakata, in Physiological and clinical aspects of short-chain fatty acids, Cambridge University Press, Cambridge, 1995 Search PubMed.
- F. P. Martin, N. Sprenger, I. Montoliu, S. Rezzi, S. Kochhar and J. K. Nicholson, J. Proteome Res., 2010, 9, 5284–5295 CrossRef CAS PubMed.
- Z. Wang, E. Klipfell, B. J. Bennett, R. Koeth, B. S. Levison, B. Dugar, A. E. Feldstein, E. B. Britt, X. Fu, Y. M. Chung, Y. Wu, P. Schauer, J. D. Smith, H. Allayee, W. H. Tang, J. A. DiDonato, A. J. Lusis and S. L. Hazen, Nature, 2011, 472, 57–63 CrossRef CAS PubMed.
- X. Cha and C. Zhou, in Biochemistry, ed. Y. Wang, People's Medical Publishing House, Beijing, 2008, pp. 120–159 Search PubMed.
- J. Xia and D. S. Wishart, Nat. Protoc., 2011, 6, 743–760 CrossRef CAS PubMed.
- L. Stryer, J. M. Berg and T. L. Tymoczko, in Biochemistry, W. H. Freeman and Company, New York, 2006 Search PubMed.
- X. Cha and C. Zhou, in Biochemistry, ed. X. Cui, People's Medical Publishing House, Beijing, 2008, pp. 220–235 Search PubMed.
- X. Zheng, G. Xie, A. Zhao, L. Zhao, C. Yao, N. H. Chiu, Z. Zhou, Y. Bao, W. Jia, J. K. Nicholson and W. Jia, J. Proteome Res., 2011, 10, 5512–5522 CrossRef CAS PubMed.
- S. S. Heinzmann, C. A. Merrifield, S. Rezzi, S. Kochhar, J. C. Lindon, E. Holmes and J. K. Nicholson, J. Proteome Res., 2012, 11, 643–655 CrossRef CAS PubMed.
- F. Savino, L. Cordisco, V. Tarasco, E. Locatelli, D. Di Gioia, R. Oggero and D. Matteuzzi, BMC Microbiol., 2011, 11, 157 CrossRef PubMed.
- B. van den Bogert, M. Meijerink, E. G. Zoetendal, J. M. Wells and M. Kleerebezem, PLoS One, 2014, 9, e114277 Search PubMed.
- H. Meng, Y. Zhang, L. Zhao, W. Zhao, C. He, C. F. Honaker, Z. Zhai, Z. Sun and P. B. Siegel, PLoS One, 2014, 9, e89862 Search PubMed.
- F. H. Karlsson, D. W. Ussery, J. Nielsen and I. A Nookaew, Microb. Ecol., 2011, 61, 473–485 CrossRef PubMed.
- D. L. Topping and P. M. Clifton, Physiol. Rev., 2001, 81, 1031–1064 CAS.
- J. M. Wong, R. de Souza, C. W. Kendall, A. Emam and D. J. Jenkins, J. Clin. Gastroenterol., 2006, 40, 235–243 CrossRef CAS PubMed.
- J. J. Kovarik, W. Tillinger, J. Hofer, M. A. Hölzl, H. Heinzl, M. D. Saemann and G. J. Zlabinger, Eur. J. Clin. Invest., 2011, 41, 291–298 CrossRef CAS PubMed.
- C. de Filippo, D. Cavalieri, M. Di Paola, M. Ramazzotti, J. B. Poullet, S. Massart, S. Collini, G. Pieraccini and P. Lionetti, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14691–14696 CrossRef PubMed.
- B. A. White, R. Lamed, E. A. Bayer and H. J. Flint, Annu. Rev. Microbiol., 2014, 68, 279–286 CrossRef CAS PubMed.
- H. Zhang, J. K. DiBaise, A. Zuccolo, D. Kudrna, M. Braidotti, Y. Yu, P. Parameswaran, M. D. Crowell, R. Wing, B. E. Rittmann and R. Krajmalnik-Brown, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2365–2370 CrossRef CAS PubMed.
- F. Brito, C. Zaltman, A. T. Carvalho, R. G. Fischer, R. Persson, A. Gustafsson and C. M. Figueredo, Eur. J. Gastroenterol. Hepatol., 2013, 25, 239–245 CrossRef PubMed.
- G. D. Wu, F. D. Bushmanc and J. D. Lewis, Anaerobe, 2013, 24, 117–120 CrossRef PubMed.
- M. Pop, A. W. Walker, J. Paulson, B. Lindsay, M. Antonio, M. A. Hossain, J. Oundo, B. Tamboura, V. Mai, I. Astrovskaya, H. Corrada Bravo, R. Rance, M. Stares, M. M. Levine, S. Panchalingam, K. Kotloff, U. N. Ikumapayi, C. Ebruke, M. Adeyemi, D. Ahmed, F. Ahmed, M. T. Alam, R. Amin, S. Siddiqui, J. B. Ochieng, E. Ouma, J. Juma, E. Mailu, R. Omore, J. G. Morris, R. F. Breiman, D. Saha, J. Parkhill, J. P. Nataro and O. C. Stine, Genome Biol., 2014, 15, R76 CrossRef PubMed.
- O. Revelles, M. Espinosa-Urgel, T. Fuhrer, U. Sauer and J. L. Ramos, J. Bacteriol., 2005, 187, 7500–7510 CrossRef CAS PubMed.
- P. Corfield, S. A. Wagner, J. R. Clamp, M. S. Kriaris and L. C. Hoskins, Infect. Immun., 1992, 60, 3971–3978 Search PubMed.
- J. C. Byrd and R. S. Bresalier, Cancer Metastasis Rev., 2004, 23, 77–99 CrossRef CAS.
- D. Monleon, J. M. Morales, A. Barrasa, J. A. López, C. Vázquez and B. Celda, NMR Biomed., 2009, 22, 342–348 CrossRef CAS PubMed.
- S. Kang, S. E. Denman, M. Morrison, Z. Yu, J. Dore, M. Leclerc and C. S. McSweeney, Inflammatory Bowel Dis., 2010, 16, 2034–2042 CrossRef PubMed.
- S. O. Noor, K. Ridgway, L. Scovell, E. K. Kemsley, E. K. Lund, C. Jamieson, I. T. Johnson and A. Narbad, BMC Gastroenterol., 2010, 12, 134 CrossRef PubMed.
- B. Samb-Ba, C. Mazenot, A. Gassama-Sow, G. Dubourg, H. Richet, P. Hugon, J. C. Lagier, D. Raoult and F. Fenollar, PLoS One, 2014, 9, e87419 Search PubMed.
- R. Cermak and G. M. Breves, Arch. Anim. Nutr., 2006, 60, 180–189 CrossRef CAS PubMed.
- J. K. icholson, E. Holmes, J. Kinross, R. Burcelin, G. Gibson, W. Jia and S. Pettersson, Science, 2012, 336, 1262–1267 CrossRef PubMed.
- T. A. Clayton, FEBS Lett., 2012, 586, 956–961 CrossRef CAS PubMed.
- F. P. Martin, Y. Wang, I. K. Yap, N. Sprenger, J. C. Lindon, S. Rezzi, S. Kochhar, E. Holmes and J. K. Nicholson, J. Proteome Res., 2009, 8, 3464–3474 CrossRef CAS PubMed.
- P. H. Yancey, M. E. Clark, S. C. Hand, R. D. Bowlus and G. N. Somero, Science, 1982, 217, 1214–1222 CAS.
- P. Kalač, Meat Sci., 2006, 73, 1–11 CrossRef PubMed.
- H. P. Til, H. E. Falke, M. K. Prinsen and M. I. Willems, Food Chem. Toxicol., 1997, 35, 337–348 CrossRef CAS.
- S. Bodmer, C. Imark and M. Kneubühl, Inflammation Res., 1999, 48, 296–300 CrossRef CAS.
- S. R. Modi, J. J. Collins and D. A. Relman, J. Clin. Invest., 2014, 124, 4212–4218 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1 shows 600 MHz 1H NMR spectra of faeces and urine of SD rat, Fig. S2 shows PCA scores scatter plots for 1H NMR data of urine and faeces, Fig. S3 shows PCA scores scatter plot for DGGE fingerprint of day 21. Tables S1 and S2 list specific assignments of faecal and urinary metabolites. Tables S3 and S4 exhibit results about quantitative enrichment analysis and pathway analysis of faecal metabolites on day 7. Tables S5 and S6 exhibit similar results like Tables S3 and S4 about urine. See DOI: 10.1039/c5ra16338b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |