
Enhanced formaldehyde selectivity in catalytic methane oxidation by vanadia on Ti-doped SBA-15†
Abstract
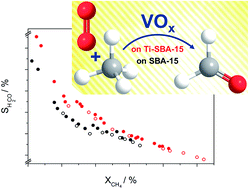
Vanadia species (2.5 wt% V) were supported on 0.2 wt% Ti-doped SBA-15 (V/Ti-SBA-15) and tested towards the selective oxidation of methane to formaldehyde. V/Ti-SBA-15 shows improved redox properties and higher selectivity towards formaldehyde over all conversions compared to VOx on pure SBA-15 (V/SBA-15).
 Please wait while we load your content...
Please wait while we load your content...

