
Direct in situ observation and explanation of lithium dendrite of commercial graphite electrodes
Zhansheng Guo*ab,
Jianyu Zhua,
Jiemin Fengbc and
Shiyu Dud
aShanghai Institute of Applied Mathematics and Mechanics, Shanghai 200072, China. E-mail: davidzsguo@shu.edu.cn; Fax: +86-21-36033287; Tel: +86-21-56331451
bShanghai Key Laboratory of Mechanics in Energy Engineering, Shanghai 200072, China
cDepartment of Mechanics, College of Science, Shanghai University, Shanghai 200444, China
dDivision of Functional Materials and Nanodevices, Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo, Zhejiang 315201, China
First published on 10th August 2015
Abstract
Lithium-ion batteries (LIBs) have some serious safety problems, such as lithium dendrite formation during charging/discharging cycles that may cause internal short-circuiting, fires, and even explosions. A new double-scale in situ experimental setup, which can record all phenomena during the electrochemical testing, was developed. Lithium dendrite growth behavior of commercial LIBs during small-current-density charging at room temperature was observed in situ. The formation, growth, and dissolution of lithium dendrites, and dead lithium residue were all observed and recorded using this new experimental test system. A detailed model of lithium electrodeposition and dissolution processes was proposed. The electrode structures were determined by X-ray diffraction (XRD). The surface morphologies were examined by scanning electron microscopy (SEM). The texture and surface morphology of the graphite active layer affected lithium dendrite initiation as well as its growth processes.
1. Introduction
Lithium-ion batteries (LIBs) are state-of-the-art power sources for portable electronic devices, electric vehicles, smart grids, tools, and toys. As the lightest and most electropositive metal, Li metal is an ideal anode material for rechargeable batteries.1,2 However, the development of secondary lithium metal batteries is hindered by the propensity of lithium to deposit in dendritic structures during battery recharging; the formation and growth of these structures compromises cell safety by creating the possibility for internal short-circuiting, catching fire, and even explosion.In the past 40 years, lithium dendrite formation during charge/discharge cycles has been widely studied through computational investigations1–9 and experimental observations.2,10–24 Arora et al.3 used a one-dimensional macro-homogeneous cell model to describe lithium deposition in a LixC6:LiyMn2O4 system. Tang et al.4 used a simplified COMSOL Multiphysics model to explain why lithium deposition occurs preferentially at electrode edges and to investigate the magnitude of edge effects. Monroe and Newman5 presented a comprehensive mathematical model of the dendrite growth process in a parallel-electrode lithium/polymer cell during galvanostatic charging. The growth model is surface-energy controlled, incorporating the effect of dendrite tip curvature into its dendrite growth kinetics. The modeling work by Akolkar6 extended Monroe and Newman's model to liquid electrolytes by incorporating the concentration dependence of the diffusion coefficient. In a subsequent paper, Akolkar7 modified his prior theoretical model6 to quantify the dendritic growth rate at sub-ambient temperature. In this model, he found that low temperature causes the increase of the lithium-ion diffusion resistance and the decrease of the surface film thickness, which are favorable conditions for the formation of dendrites during lithium electrodeposition. Recently, Aryanfar et al.8 studied dendrite nucleation and growth on the basis of a scaled coin cell prototype fitted with a lithium metal electrode. Many theoretical models have been proposed in order to gain deep insights into the physics of the formation and growth process of Li dendrite, though these models are not based on the popular graphite electrode but on the lithium metal electrode.
For safe operation of battery systems, the experimental observation of Li dendrite evolution during cycling must be obtained. For this purpose, many imaging studies have been performed on the morphology of lithium depositions, which have included optical microscopy,10–17 scanning electron microscopy (SEM),12,16,18–20 transmission electron microscopy (TEM),11,12,21–24 atomic force microscopy (AFM),18,25,26 nuclear magnetic resonance spectroscopy (NMR),11,12,26–30 magnetic resonance imaging (MRI),11,12,31 electrochemical impedance spectroscopy (EIS),18,20 neutron diffraction,32 Raman spectroscopy,33 and X-ray diffraction (XRD).34 Since the 1990s, SEM, TEM, and AFM have been extensively used for in situ and ex situ research on Li deposition for their capability of providing visual evidence of the morphology, and high resolutions have been achieved. Shao12 reviewed the roles of microscopic techniques (X-ray fluorescence (XRF) microscopy, Raman microscopy, transmission X-ray microscopy (TXM), SEM, and TEM) in real-time monitoring of the dynamic changes in the LIB electrodes and materials. Ramdon et al.25 presented a review of in situ electrochemical cells using AFM. They studied the morphological information of LiFePO4 cathodes during charge/discharge cycles. Arai et al.27 observed lithium metal deposition during overcharge in LIBs by in situ solid state 7Li NMR. Recently, Harks et al.11 gave a comprehensive review of techniques applied for LIB research, where they discussed and described in situ techniques, cell design, as well as some research reports. Although various methods have been used to study electrodeposition in order to understand the mechanisms involved, they have generally provided only qualitative information about the amount and type of microstructure. Most of these experimental techniques are also destructive.
We have found that theoretical studies and experimental observation of Li dendrites mostly focused on half-cells with lithium metal as the positive electrode. The operating conditions usually involve low temperature or high charge rates. However, the problem of lithium dendrite growth of today's popular commercial graphite electrodes has not been addressed, since there is currently not enough research being conducted, especially for common low charge rates at room temperature. Several researchers10–17 have studied the Li dendrites on commercial electrodes using non-destructive techniques. Unfortunately, their spatial scales are very small and a lot of information could not be obtained. In this study, we developed a double-scale experimental setup for direct in situ observation of the phenomena of commercial full cells under real operating conditions (small current charge/discharge at room temperature). Graphite/LiFePO4 materials were selected to investigate the lithium dendrite deposition/dissolution process during galvanostatic charge/discharge cycles. The structures of the studied graphite electrodes were determined by X-ray diffraction (XRD). The surface morphologies were examined by scanning electron microscopy (SEM). These in situ direct and non-destructive observation results will provide insight into the mechanism of lithium electrodeposition and dissolution of practical commercial LIBs.
2. Experimental
2.1 Electrode materials and size
Note that nearly all LIBs currently in the market are based on graphite anodes. Two types of graphite electrodes were tested. The thickness of the negative electrodes was 100 μm. The thickness of the active layer (i.e., graphite) was 91 μm and the thickness of the copper current collector was 9 μm. The LiFePO4 electrodes were used to serve as the metal oxide cathode. All electrodes were dried overnight at room temperature and stored in an oven at 90 °C before use. Celgard 2325 (25 μm thickness, Celgard, USA) was used as a separator. The commercial electrolyte (1 M LiPF6 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio mixture of ethylene carbonate and dimethyl carbonate) was used in all test cells.
:1 weight ratio mixture of ethylene carbonate and dimethyl carbonate) was used in all test cells.
The graphite electrodes used in this study were 8 mm long and 4 mm wide. The LiFePO4 positive electrode was completely packed by a separator so that the cell would not undergo internal short circuits during the lithium dendrite growth. The distance between the positive and negative electrode was 1–2 mm.
2.2 Cell design and assembly
The electrodes were placed side-by-side in order to allow clear observation as shown in Fig. 1. This arrangement of the electrode structure may not be sufficient to fully understand the mechanisms of lithium dendrite growth for a real battery; nevertheless, it can at least provide useful clues to the process controlling the growth of lithium deposits. They were verified by the observation of lithium dendrites in a real commercial battery.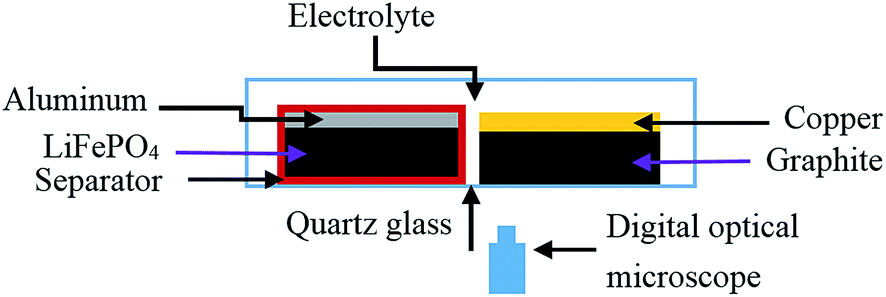 | ||
| Fig. 1 Schematic of the electrochemical cell for the in situ imaging of lithium dendrite changes. | ||
All tested cells were assembled or disassembled in an argon-filled glove box (O2 < 1 ppm, H2O < 0.1 ppm, Mikrouna, China).
2.3 Electrochemical testing
A BK-6808 battery test system (Guangzhou Blue-Key Electronic Industry Co., Ltd, China) was used to galvanostatic charge/discharge cycles of our cells with a current of 0.2C (that is to say, the battery would be fully charged within 300 min). The data acquisition frequency was 1 Hz. A relaxation period of 10 min with open-circuit potential followed each charging and discharging step. All of the electrochemical tests were performed at room temperature. For the real commercial LIBs, the charged constant current was 1C (the battery would be fully charged within 60 min) at room temperature.2.4 Double-scale in situ experimental observation system
In order to observe the lithium dendrite growth process of negative electrodes in situ during charging/discharging cycles, a new double-scale (micro-scale and macro-scale) non-destructive experimental test system was developed. The schematic of the setup is shown in Fig. 1. The boxes of the newly designed cells were all made of quartz glass (2 mm thickness, Baosheng Quartz Products Co., Ltd, China). The real-time phenomena of electrodes occurring during charging/discharging cycles, such as the lithium dendrite growth process, gas generation, lithium ion transport path (judged by color changes for the graphite electrode), deformation, and delamination, were all recorded directly by this fully transparent cell from all directions. The micro-scale in situ observation was realized by digital optical microscopy (DOM, Huabai H800X, China) and macro-scale observation was performed by naked eyes or other low-resolution CCD cameras. It should be pointed out that the structure of the cell can be assembled to be flexible according to the objective of investigations. However, in this paper, we will focus on lithium dendrite growth processes of commercially popular negative electrodes. While the electrochemical measurements were performed, video sequences of lithium electrodeposition/dissolution were recorded by DOM. Based on our several pre-test results, the image acquisition time for DOM was set to 1 min. The advantage of this new double-scale experimental equipment over other single-scale (micro-scale or macro-scale) observation systems will be discussed in the following section.3. Results and discussion
3.1 Lithium electrodeposition of graphite sample A/LiFePO4 battery
In this paper, we define the overcharge time as the time after a 100% theoretical state of charge (SOC) was reached during constant current charging. Fig. 2(a–f) shows several individual frames extracted from the in situ video sequence. These show the variation of lithium dendrites of graphite sample A during the first charge. Fig. 2(a) shows that the metallic lithium (most moss-like) was deposited on the surface of the end of the graphite electrode when the overcharge time was only 10 min (as indicated by the marked region). The marked region is enlarged in Fig. 2(e) for a clearer view. Lithium metal deposited on the negative electrode may react quickly with solvent molecules or solute in the vicinity, yielding Li2CO3, LiF, or other insoluble products.33 The nature of the final product is not our major concern in this paper; rather, the major issue is that lithium is irreversibly lost. We can clearly see lithium dendrite deposition on the surface of the electrode when the theoretical overcharge time is 34 min, which is shown in the marked region of Fig. 2(b). When overcharging of the electrode continued for 60 min, the Li dendrites were found to increase in thickness and grow in a straight line, as can be seen by comparing Fig. 2(b) and (c). During this process, the electrode deposition morphology seemed unchanged. This is because a solid electrolyte interface (SEI), which is undetectable owing to the spatial resolution limits of the present DOM images, will protect the metallic lithium from further reacting with the electrolyte.35 In the next stage with the overcharge time increased to 100 min, when lithium insertion sites multiplied and became simultaneously active, the dendrites began to grow in width, and significant protuberances became visible as shown in Fig. 2(d) (as indicated by arrows). Fig. 2(d) and the corresponding partial enlargement in Fig. 2(f) show that the protuberances are surrounded by the dendrites and that the area of the dendrites also increased. Based on the basic laws of the evolution of crystalline structures, it is reasonable to assume that this bush-shaped metallic lithium could have been nearly linear and needle-like at the initial stage of lithium dendrite formation and grew to the current shape exhibited in the image at the end of stage. It is interesting that lithium dendrite was observed at the 0.2C charge rate, which is consistent with theoretical calculations.5 It has been found that dendrite growth can occur even if operating at well below the limiting current density. This will be explained more clearly after the structure and surface morphology of the electrode are investigated (see XRD and SEM studies). | ||
| Fig. 2 In situ observation of formation of lithium dendrites on graphite sample A during first charge: (a) dendrite region found; (b) dendrite clearly observable; (c) moss-like dendrite formed; (d) protuberance structure; (e) zoomed image of the marked area in (a); (f) zoomed image of the marked area in (d). | ||
It should be noted that other phenomena are observed through this double-scale experimental observation system during charging/discharging cycles. First, many bubbles appeared during the first charge, and they completely disappeared after several charge/discharge cycles. Bubbles are the products of side reactions during the generation of the SEI film, and their main components are CO2 and C2H4.36,37 Second, color changes that indicate the lithium ion transport path during charging and discharging were also clearly observed. Harris et al.17 studied this phenomenon in detail. These images confirm that this in situ macro-micro observation setup can be used to not only study the mechanism of lithium dendrite growth but also measure the lithium ion transport directly. Lastly, the center region of this graphite electrode cannot be fully charged (i.e., do not transform into a golden color), even when all other locations have been shown to be fully charged (golden color). The cells assembled with graphite sample A all display this phenomenon. Further study is in progress to determine the reason for this behavior.
3.2 Lithium dissolution of graphite sample A/LiFePO4 battery
Fig. 3(a–d) show the dissolution of lithium dendrites of graphite sample A during discharge. Comparison between the marked region of Fig. 3(a) and (b) showed that the dissolution process began at the protuberances. Comparing Fig. 3(c) with Fig. 3(b) revealed that the protuberances then gradually dissolved while the dendrite area decreased (the color of the dendrite became lighter and the areas became smaller), and a thin moss-shaped shell, marked by the dashed circles, remained, as indicated by the difference between Fig. 3(c) and (d). From the video (not shown here), it was found that most dendrite dissolution occurred when the depth of discharge (DOD, the ratio between the discharge capacity and total capacity) was only approximately 18%. The dissolution of lithium dendrite is much faster at the initial stage of discharge than at the later stage of discharge, which can be seen by comparing the discharge times of Fig. 3(a) and (b) and Fig. 3(b) and (c). Comparison between Fig. 3(c) and (d) revealed that there was no clear difference in dendritic morphology. That is to say, dissolution of Li dendrites was nearly complete after a discharge time of 48 min. Fig. 3(a–d) show that most of the lithium dendrite can be dissolved. The irreversible lithium dendrite (the so-called dead lithium) was still present on the electrode after discharging had ended (Fig. 3(d)). It should be noted that some dead lithium, which increases the surface roughness of the electrode and causes enhanced lithium growth during the deposition in the second charge/discharge cycle, is attached to the electrode permanently. The lithium deposits, which are no longer attached to the electrode and do not participate in the subsequent charge/discharge cycle, were also found in all of our experiments (not shown here). | ||
| Fig. 3 In situ observation of dissolution of lithium dendrites on graphite sample A during first discharge: (a) original state before discharging; (b) state after 10 min of discharge; (c) state after 48 min of discharge; (d) state after full discharge. | ||
3.3 Lithium electrodeposition of a real commercial graphite/LiFePO4 battery
As we mentioned in the experimental section, the arrangement of electrodes is different with real LIBs, in which electrodes are usually spirally wound or stacked. In order to compare our design with those of real batteries, we repeated experiments using real serviced commercial graphite electrode LIBs. Fig. 4 shows some of the results when the LIBs are disassembled under controlled and uncontrolled environmental conditions. They will be described and analyzed in detail in our forthcoming publication. Here, we give only a very brief description of the results. Many different lengths of needle-like lithium dendrites can be seen clearly in Fig. 4(a). Some bush-like lithium dendrites, which most likely form from the needle-like dendrites, were also observed. The results indicate that dendrites do not begin growing simultaneously, because the lithium depositions are not uniform. This might also suggest heterogeneities of the current density or the existence of strong local electric fields. Fig. 4(b) shows the real LIBs, which catch fire when disassembled under air conditions. It is reasonable to conclude that such lithium dendrites will pose serious problems both in terms of performance and safety of LIBs. | ||
| Fig. 4 Lithium dendrites on a graphite electrode in a real serviced commercial battery: (a) graphite electrode disassembled under an argon environment; (b) graphite electrode disassembled under an air environment. | ||
It must be pointed out that this real battery is charged galvanostatically with a 1C charge rate at room temperature. The SOC begins at 0% and ends theoretically at 100%. That is to say, the battery does not overcharge. Based on Fig. 4(a), it is reasonable to believe that some needle-like lithium depositions probably formed at far below the theoretical full SOC.
3.4 Process of lithium electrodeposition and dissolution
In conjunction with the aforementioned experimental studies (Fig. 2–4), an ideal model to describe lithium electrodeposition and dissolution processes on commercial graphite electrodes is suggested in this work. Fig. 5 shows the details of the model to explain the observations from our experiments. Fig. 5(a) shows an ideal structure of the graphite electrode before Li deposition occurs. The surfaces of graphite intercalation compounds (GICs) are covered completely by SEIs. However, an SEI film will rupture at some specific locations where lithium metal is stochastically nucleated (such as points 1 and 2 in Fig. 5(b)) during the lithiation process. Lithium deposits will enhance SEI film cracking, which in turn accelerates the rate of lithium formation in these areas, resulting in the generation of lithium dendrites. It is most likely that the nucleated Li metal will grow into a whisker-like structure first, and then form needle-like dendrites by gaining in thickness and growing in a straight line (points 1 and 2 in Fig. 5(b)). At the same time, the formation of new lithium ions will occur randomly (point 3 in Fig. 5(c)), and lateral growth is also possible (point 1 in Fig. 5(c)) because the current density may be concentrated at local protrusions during lithium electrodeposition. Li metal can be electrodeposited not only on the tip (point 4 in Fig. 5(d)) but also near the foot of the dendrite part embedded in the SEI film (point 5 between 2 and 3 in Fig. 5(d)). Some whisker dendrites aggregate with adjacent dendrites to form massive dendrites, as illustrated in Fig. 5(e) and Fig. 2(f). | ||
| Fig. 5 Schematic of suggested morphology evolution of the graphite electrode during lithium electrodeposition and dissolution: (a) original state at full charge; (b) random nucleation of Li metal during overcharge; (c) dendrite growth and formation of new nucleated lithium; (d) continuation of dendrite growth and formation of new nucleated lithium between 2 and 3; (e) formation of moss-like dendrite; (f) state of dendrite at the first stage of discharge; (g) state of dendrite at middle stage of discharge; (h) state of dendrite at the end of discharge. | ||
At the initial stage of lithium discharge, lithium dendrites of all shapes (moss-like or needle-like) begin to shrink, as revealed by the comparison between Fig. 5(e) and (f). Then, the lengths and thicknesses of needle-like dendrites will decrease (Fig. 5(g)), and moss-like dendrites also shrink during discharge. After full discharge, most dendrites dissolved. Therefore, lithium dissolution is almost an opposite process to deposition, except that dead lithium resides on and contacts with the graphite electrode through an SEI.35 It should be noted that a small amount of dead lithium, which disconnects from the electrode and moves into the electrolyte, was also observed in our direct in situ double-scale experiments (which is not shown in Fig. 5).
3.5 Lithium electrodeposition and dissolution of graphite sample B/LiFePO4 battery
The images of cells assembled using graphite sample B as the anode during charge/discharge cycles are shown in Fig. 6. The cells were assembled in the same manner as the previous ones except for the negative electrode.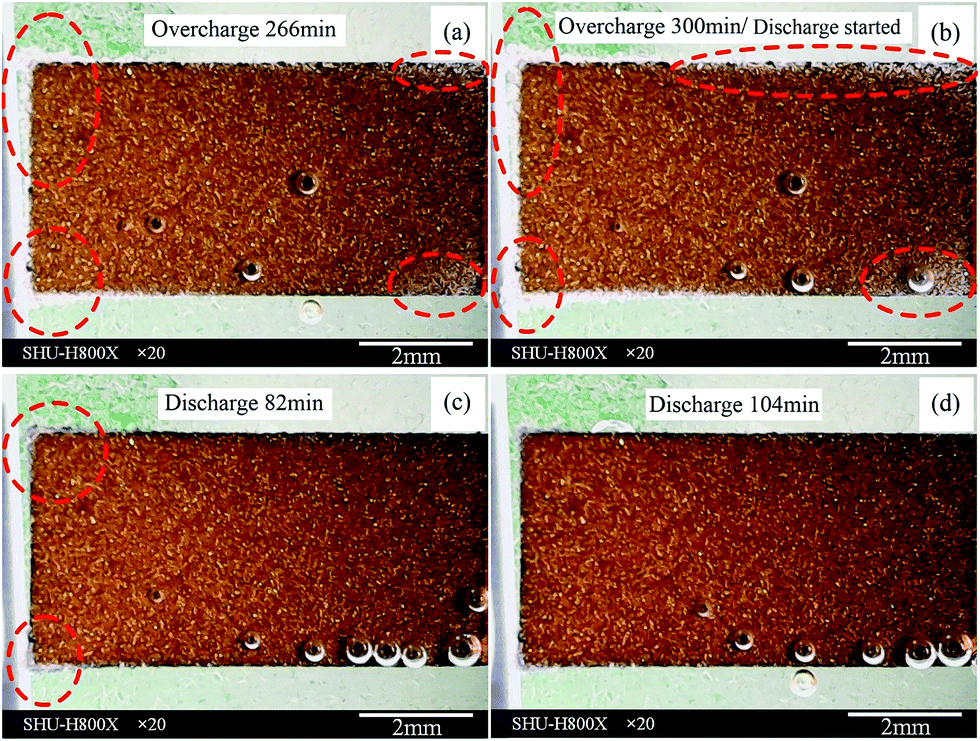 | ||
| Fig. 6 In situ observation of lithium dendrites on graphite sample B during first electrochemical cycle: (a) state of dendrite found; (b) original state before discharge; (c) state after 82 min of discharge; (d) state after 104 min of discharge. | ||
It should be noted that we could not find lithium dendrite after overcharging for 100 min, while a large amount of dendrite had been observed clearly for sample A. Thus, we overcharged this cell until dendrite could be observed by DOM. Lithium deposition was found at the edge of the graphite electrode after an overcharge time of 266 min (region marked by a dashed circle in Fig. 6(a)). The lithium deposition growth became more clearly observable and the area where lithium depositions appeared also grew (region marked by dashed circle in Fig. 6(b)) as the overcharge time increased. The deposited Li can grow in thickness, which can be observed in the right region of Fig. 6(b). This implies that the electric field direction has little effect on the lithium deposition growth direction, which agrees well with previous work by Steiger et al.15,16 From the dendrite formation time, we can see that initiation of Li deposition on graphite sample B occurred much later than that on sample A upon comparing Fig. 2(a–b) with Fig. 6(a–b). This may be because of the structures and particle sizes of these two graphite electrodes. They will be analyzed in detail by XRD and SEM, respectively. It should be pointed out that the lithium ion distribution of sample B is almost uniform, as the color of the graphite electrode is almost the same over the entire surface of the electrode.
During the lithium dissolution, the lithium deposition at the right region disappeared completely, as revealed by comparison between Fig. 6(b) and (c). This indicates that the lithium deposits at these locations are more easily dissolved. All lithium deposits were dissolved completely after a 104 min discharge time, with a DOD of approximately 34%, with no dead lithium residing on the graphite electrode after full discharge. It can be concluded that the cell assembled using graphite sample B undergoes little capacity loss owing to lithium electrodeposition. This graphite electrode is much better than the previous (sample A) for practical usage as a commercial negative electrode. It seems that different graphite electrodes have significant effects on Li dendrite deposition and cell performance. It has been noted that the lithium dissolution time of graphite sample B is also much longer than that of graphite sample A. This phenomenon is still not fully understood.
3.6 Electrode structures and surface morphologies of used graphite samples
In order to determine the likely reason for the very different behaviors of these two graphite electrode samples, XRD (18 kW D/MAX2550, Rigaku, Japan) using CuKα radiation was employed to identify the physical properties of the graphite samples and SEM (HITACHI SU-1500, Japan) experiments were performed to observe the surface morphologies. Fig. 7 shows XRD patterns of sample A and sample B. The standard spectra of the graphite hexagonal phase (2H, XYXY stacked structure) are also shown in image (c). Fig. 7(a) and (b) reveal that they clearly show all four peaks. By comparing with the standard pattern of graphite 2H (Fig. 7(c)), it can be confirmed that these two different graphite samples are graphite 2H. Notably, the (0 0 2) peak intensity of sample A (Fig. 7(a)) is much stronger than that of sample B (Fig. 7(b)). Quantitative analysis of these XRD patterns indicates 75% and 64% preferential orientation of (0 0 2) peaks for samples A and B, respectively. That is to say, the graphitization of sample A is better than that of sample B. Fig. 7 also shows the magnified pattern at XRD angles of 40°–60°. The peaks at ca. 42.3° and 44.4° are attributable to the (1 0 0) and (1 0 1) planes of the hexagonal structure, respectively. The (1 0 1) plane of rhombohedral (3R, XYZXYZ stacked structure) graphite appeared at ca. 43.5°. This indicates that samples also show different contents of the rhombohedral phase. According to the work of Simon et al.,38 the rhombohedral content by itself has no significant influence on the capacity or electrochemical potential but has a remarkable impact on irreversibility losses. Combined with our in situ direct experimental observations of lithium electrodeposition and dissolution processes, we believed that lithium intercalation is influenced by the formation of lithium dendrite though quantification of structural effects is difficult mainly owing to complications caused by multiple factors (graphitization, density of dislocation, impurities, particle shape and size, etc.). | ||
| Fig. 7 X-ray diffraction patterns of (a) sample A and (b) sample B, and (c) standard spectra of graphite 2H. | ||
Fig. 8 shows the SEM images of these two graphite samples. Fig. 8(a) shows graphite sample A fabricated by flake or plate structures. The particle sizes varied from 10 μm to 40 μm. These particles show an unconsolidated arrangement, which leads to the formation of an uneven surface and high porosity. These will affect not only the electrochemical properties but also the lithium deposition on the surface of graphite sample A. Fig. 8(b) shows graphite sample B fabricated from many spherical particles. The diameters of these particles vary from 2 μm to 40 μm. It can be observed clearly that the surface of sample B is denser than that of graphite sample A. Small particles fill the gaps between the larger ones so that the degree of surface roughness seems smaller and the porosity is lower. This provides sample B with a more uniform current, since current distribution is more uniform on flat smooth surfaces. According to ref. 1 and 2, lithium deposits occur only at locations in the anode where the overpotential falls below zero vs. Li/Li+, which is caused by non-uniform current distribution. From our in situ direct experimental observations, we confirmed that graphite sample A does generate lithium dendrite more easily than does graphite sample B. It is highly probable that rough surfaces and porosity enhance the local current density (current localization), promoting dendrite growth in those specific regions. Our results also provided some confirmation for speculation by Harris and Lu39 that heterogeneity was related to lithium dendrite formation. Since the graphite surface roughness and porosity depend on the particle size distributions, one can deduce that particles used to fabricate an electrode with a broader distribution of size will have a more inhibitory effect on lithium dendrites than those with uniform size distributions.
 | ||
| Fig. 8 SEM images of (a) graphite sample A and (b) graphite sample B. | ||
4. Conclusions
To better understand lithium deposition growth and dissolution, the lithium dendrite growth process was investigated by using a newly designed double-scale in situ experimental observation system, which was successfully tested. All phenomena (color change, gas generation, lithium dendrite growth, deformation, etc.) that occurred during charging/discharging cycles were observed. The formation, growth, and dissolution of lithium dendrites, and dead lithium residues of two different commercial graphite electrodes were observed. The dendrites can occur at some locations where the state-of-charge is far below 100% as a result of improper graphite electrode selection. The structural factors of graphite anodes for the reversible dissolution and deposition of lithium dendrite during charging/discharging cycles were proposed. Most dead lithium was still attached to the electrode after full discharge. Lithium dendrite initiation and its growth processes are affected by the texture, structure, and degree of surface roughness of graphite electrodes.Acknowledgements
The authors gratefully acknowledge the financial support by the National Science Foundation of China (No. 11472165 and 11332005).References
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 CAS.
- Z. Li, J. Huang, B. Y. Liaw, V. Metzler and J. B. Zhang, J. Power Sources, 2014, 254, 168–182 CrossRef CAS PubMed.
- P. Arora, M. Doyle and R. E. White, J. Electrochem. Soc., 1999, 146, 3543–3553 CrossRef CAS PubMed.
- M. Tang, P. Albertus and J. Newman, J. Electrochem. Soc., 2009, 156, A390–A399 CrossRef CAS PubMed.
- C. Monroe and J. Newman, J. Electrochem. Soc., 2003, 150, A1377–A1384 CrossRef CAS PubMed.
- R. Akolkar, J. Power Sources, 2013, 232, 23–28 CrossRef CAS PubMed.
- R. Akolkar, J. Power Sources, 2014, 246, 84–89 CrossRef CAS PubMed.
- A. Aryanfar, D. Brooks, B. V. Merinov, W. A. Goddard, A. J. Colussi and M. R. Hoffmann, J. Phys. Chem. Lett., 2014, 5, 1721–1726 CrossRef CAS.
- R. D. Perkins, A. V. Randall, X. Zhang and G. L. Plett, J. Power Sources, 2012, 209, 318–325 CrossRef CAS PubMed.
- C. Brissot, M. Rosso, J. N. Chazalviel, P. Baudry and S. Lascaud, Electrochim. Acta, 1998, 43, 1569–1574 CrossRef CAS.
- P. P. R. M. L. Harks, F. M. Mulder and P. H. L. Notten, J. Power Sources, 2015, 288, 92–105 CrossRef CAS PubMed.
- M. Shao, J. Power Sources, 2014, 270, 475–486 CrossRef CAS PubMed.
- J. Wandt, C. Marino, H. A. Gasteiger, P. Jakes, R.-A. Eichel and J. Granwehr, Energy Environ. Sci., 2015, 8, 1358–1367 CAS.
- K. Nishikawa, T. Mori, T. Nishida, Y. Fukunaka, M. Rosso and T. Homma, J. Electrochem. Soc., 2010, 157, A1212–A1217 CrossRef CAS PubMed.
- J. Steiger, D. Kramer and R. Mönig, Electrochim. Acta, 2014, 136, 529–536 CrossRef CAS PubMed.
- J. Steiger, D. Kramer and R. Mönig, J. Power Sources, 2014, 261, 112–119 CrossRef CAS PubMed.
- S. J. Harris, A. Timmons, D. R. Baker and C. Monroe, Chem. Phys. Lett., 2010, 485, 265–274 CrossRef CAS PubMed.
- N. Schweikert, A. Hofmann, M. Schulz, M. Scheuermann, S. T. Boles, T. Hanemann, H. Hahn and S. Indris, J. Power Sources, 2013, 228, 237–243 CrossRef CAS PubMed.
- F. Orsini, A. D. Pasquier, B. Beaudoin, J. M. Tarascon, M. Trentin, N. Langenhuizen, E. D. Beer and P. Notten, J. Power Sources, 1999, 76, 19–29 CrossRef.
- G. Bieker, M. Winter and P. Bieker, Phys. Chem. Chem. Phys., 2015, 17, 8670–8679 RSC.
- X. H. Liu and J. Y. Huang, Energy Environ. Sci., 2014, 4, 3844–3860 Search PubMed.
- X. H. Liu, L. Zhong, L. Q. Zhang, A. Kushima, S. X. Mao, J. Li, Z. Z. Ye, J. P. Sullivan and J. Y. Huang, Appl. Phys. Lett., 2011, 98, 183107 CrossRef PubMed.
- H. Ghassemi, M. Au, N. Chen, P. A. Heiden and R. S. Yassar, Appl. Phys. Lett., 2011, 99, 123113 Search PubMed.
- R. L. Sacci, N. J. Dudney, K. L. More, L. R. Parent, I. Arsian, N. D. Browning and R. R. Unocic, Chem. Commun., 2014, 50, 2104–2107 RSC.
- S. Ramdon, B. Bhushan and S. C. Nagpure, J. Power Sources, 2014, 249, 373–384 CrossRef CAS PubMed.
- R. Bhattacharyya, B. Key, H. L. Chen, A. S. Best, A. F. Hollenkamp and C. P. Grey, Nat. Mater., 2010, 9, 504–510 CrossRef CAS PubMed.
- J. Arai, Y. Okada, T. Sugiyama, M. Izuka, K. Gotoh and K. Takeda, J. Electrochem. Soc., 2015, 162, A952–A958 CrossRef CAS PubMed.
- R. E. Gerald, C. S. Johnson, J. W. Rathke, R. J. Klingler, G. Sandi and L. G. Scanlon, J. Power Sources, 2000, 89, 237–243 CrossRef CAS.
- K. Gotoh, M. Izuka, J. Arai, Y. Okada, T. Sugiyama, K. Takeda and H. Ishida, Carbon, 2014, 79, 380–387 CrossRef CAS PubMed.
- M. Letellier, F. Chevallier and M. Morcrette, Carbon, 2007, 45, 1025–1034 CrossRef CAS PubMed.
- S. Chandrashekar, N. M. Trease, H. J. Chang, L.-S. Du, C. P. Grey and A. Jerschow, Nat. Mater., 2012, 11, 311–315 CrossRef CAS PubMed.
- N. Sharma and V. K. Peterson, Electrochim. Acta, 2013, 101, 79–85 CrossRef CAS PubMed.
- S. Hy, F. Felix, Y. H. Chen, J. Liu, J. Rick and B.-J. Hwang, J. Power Sources, 2014, 256, 324–328 CrossRef CAS PubMed.
- C.-K. Lin, Y. Ren, K. Amine, Y. Qin and Z. H. Chen, J. Power Sources, 2013, 230, 32–37 CrossRef CAS PubMed.
- M. Ziera, F. Scheibac, S. Oswald, J. Thomas, D. Goers, T. Scherer, M. Klose, H. Ehrenberg and J. Eckert, J. Power Sources, 2014, 266, 198–207 CrossRef PubMed.
- R. Dedryvère, H. Martinez, S. Leroy, D. Lemordant, F. Bonhomme, P. Biensan and D. Gonbeau, J. Power Sources, 2007, 174, 462–468 CrossRef PubMed.
- D. Aurbach and M. Mashkovich, J. Electrochem. Soc., 1998, 145, A2629 CrossRef PubMed.
- B. Simon, S. Flandrois, K. Guerin, A. Fevrier-Bouvier, I. Teulat and P. Biensan, J. Power Sources, 1999, 81–82, 312–316 CrossRef CAS.
- S. J. Harris and P. Lu, J. Phys. Chem. C, 2013, 117, 6481–6492 CAS.
| This journal is © The Royal Society of Chemistry 2015 |