
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A bonding evolution theory study of the mechanism of [3+2] cycloaddition reactions of nitrones with electron-deficient ethylenes†
Mar Ríos-Gutiérreza,
Patricia Pérezb and
Luis R. Domingo*a
aUniversidad de Valencia, Facultad de Química, Departamento de Química Orgánica, Dr. Moliner 50, E-46100 Burjassot, Valencia, Spain. E-mail: domingo@utopia.uv.es; Web: http://www.luisrdomingo.com
bUniversidad Andrés Bello, Facultad de Ciencias Exactas, Departamento de Ciencias Químicas, Av. República 230, 8370146, Santiago, Chile
First published on 25th June 2015
Abstract
The mechanism of zw-type [3+2] cycloaddition (32CA) reactions of nitrones with electron-deficient (ED) ethylenes has been studied using density functional theory (DFT) methods at the MPWB1K/6-31G(d) level of theory. An exploration of the potential energy surfaces associated with the four competitive reactive channels of the 32CA reaction between C-phenyl-N-methyl nitrone and acrolein indicates that the cycloaddition reaction takes place through a one-step mechanism. This cycloaddition reaction presents a moderate meta regioselectivity and a complete endo stereoselectivity, which is diminished in dichloromethane. Analysis of the DFT reactivity indices of the reagents allows an explanation of the participation of nucleophilic nitrones in zw-type 32CA reactions towards ED ethylenes. A bonding evolution theory (BET) study of the two endo regioisomeric reactive channels allows establishing the molecular mechanism of these relevant 32CA reactions. Both regioisomeric channels topologically take place along eight differentiated phases. While the formation of the C–C single bond follows Domingo's recently proposed model, the formation of the O–C single bond takes place at the short distance of 1.6 Å through the donation of some electron density of the oxygen lone pairs of the nitrone to the β-conjugated carbon atom of acrolein. BET supports the non-concerted nature of these zw-type 32CA reactions and makes it possible to reject the pericyclic mechanism proposed for them.
Introduction
The [3+2] cycloaddition (32CA) reaction of nitrones with alkenes is one of the most interesting methods for the synthesis of isoxazolidines,1 which are important precursors for the synthesis of a large number of biologically active molecules with a diversity of applications, mainly as antiviral agents.2 The use of non-symmetric alkenes opens up the possibility of the formation of two isomeric isoxazolidines (see Scheme 1). | ||
| Scheme 1 32CA reactions of nitrones. | ||
Depending on the electronic nature of the three-atom-components (TACs) involved in the 32CA reactions, these cycloadditions have recently been classified into pseudodiradical-type (pr-type) reactions involving TACs with a high pseudodiradical character, e.g. carbonyl ylides, which take place easily through an earlier TS with non-polar character, and zwitterionic-type (zw-type) reactions involving TACs with a high zwitterionic character, e.g. nitrones, characterised by favourable nucleophilic/electrophilic interactions, taking place through polar transition state structures (TSs).3
A recent study of a series of the most common TACs used in organic synthesis,4 including nitrile ylide 1, nitrile imine 2, nitrile oxide 3, diazoalkane 4, azide 5, nitrone 6 and methyl nitronate 7, has shown that they present a high nucleophilic character, participating in zw-type reactions towards electron-deficient (ED) ethylenes (see Scheme 2).5
 | ||
| Scheme 2 TACs used in organic synthesis.5 | ||
Thus, the simplest nitrone 6 has a nucleophilicity index of N = 2.95 eV,5 being on the borderline of strong nucleophiles.6 Computed activation energies indicated that these non-substituted TACs react quickly towards the ED 1,1-dicyanoethylene (DCE) 8. Interestingly, the most favourable regioisomeric TS associated with the 32CA of nitrone 6 with ED ethylene 8, TS1-m, was found to be 1.0 kcal mol−1 below that of the separated reagents (see Scheme 3). This 32CA reaction takes place with a high global electron density transfer (GEDT),7 0.26e, in clear agreement with a zw-type 32CA reaction,3 compared with the non-polar 32CA reaction between nitrone 6 and ethylene 11 presenting an activation energy of 13.1 kcal mol−1 and a GEDT of 0.01 eV.5
 | ||
| Scheme 3 32CA reaction between the simplest nitrone 6 and DCE 8. B3LYP/6-31G(d) relative energies are given in parentheses.5 | ||
Most 32CA reactions of nitrones take place via a one-step mechanism in which the two new single bonds are formed in a more or less asynchronous manner. In addition, due to the non-symmetry of the ethylene component, two regioisomeric channels are feasible, each one of them being initialised by the formation of the O–C or the C–C single bond. As the formation pattern of the O–C and C–C single bonds along the two regioisomeric channels is different, it is expected that these channels have a dissimilar molecular mechanism.
There is growing interest in explaining chemical reactivity arising from the redistribution of the electron density along the corresponding reaction pathway. The advantage of this selection is based on the fact the electron density is a local function defined within the exact many body theory, and it is also an experimentally accessible scalar field, allowing a sound description of bonding changes to characterise a reaction mechanism8 To perform these analyses quantitatively, the bonding evolution theory (BET), consisting of the joint-use of the electron localisation function (ELF) of Becke and Edgecombe9 and Thom's catastrophe theory10 (CT), was proposed11 as a new tool for analysing the electronic changes in chemical processes. BET has been applied to different elementary reactions, allowing the molecular mechanism to be established.12
Very recently, Merino et al. studied the mechanism of the 32CA reaction between nitrone 12 and lithium ynolate 13 using DFT methods (see Scheme 4).13 An ELF topological analysis of selected points of the IRC associated with this 32CA reaction showed that after the formation of a starting molecular complex MC2-o without energy barrier, in which the lithium atom is coordinated to both nitrone 12 and ynolate 13, the 32CA reaction begins with the initial formation of the C–C single bond via a two-stage one-step mechanism,14 the activation energy being 14.7 kcal mol−1. Along this non-concerted process, the O–C single bond formation takes place at the end of the reaction path, when the first C–C single bond has been completely formed.13
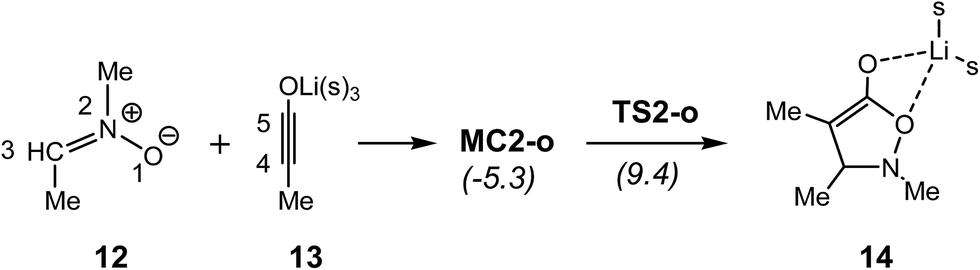 | ||
| Scheme 4 Reaction between nitrone 12 and ynolate 13. B3LYP/6-31G(d) relative energies are given in parentheses.13 | ||
Herein, a DFT study of the zw-type 32CA reaction of nitrone 15 with acrolein 16 yielding isoxazolidines 17 and/or 18, as a model of 32CA reactions of nitrones with ED ethylenes, is carried out in order to characterise the molecular mechanism of this significant class of 32CA reactions (see Scheme 5). A BET study of the two competitive endo regioisomeric reaction channels of the 32CA reaction between nitrone 15 and acrolein 16 is performed, characterising the molecular mechanism of these CA reactions.
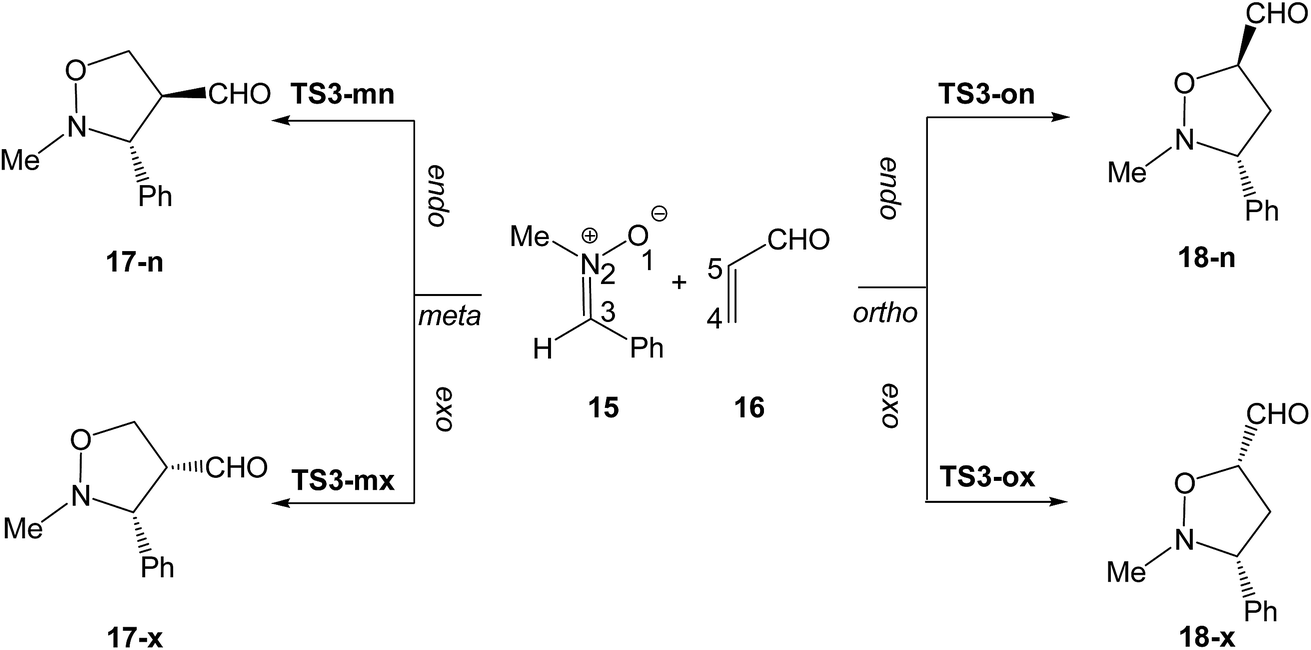 | ||
| Scheme 5 32CA reaction of nitrone 15 with acrolein 16. | ||
Computational methods
Several works have shown that the B3LYP functional15 is relatively accurate for kinetic data, although the reaction exothermicities are underestimated.16 Truhlar's group has proposed some functionals, such as the MPWB1K,17 which improve thermodynamic calculations. Therefore, in this study the MPWB1K functional was selected together with the standard 6-31G(d) basis set.18 Optimisations were performed using the Berny analytical gradient optimisation method.19 The stationary points were characterised by frequency computations in order to verify that TSs have one and only one imaginary frequency. The IRC paths20 were traced in order to check the energy profiles connecting each TS to the two associated minima of the proposed mechanism using the second order González–Schlegel integration method.21 Solvent effects of dichloromethane (DCM) in the single-point energy calculations were taken into account using the polarisable continuum model (PCM) developed by Tomasi's group22 in the framework of the self-consistent reaction field (SCRF).23 The electronic structures of the stationary points were analysed by the natural bond orbital (NBO) method24 and by the ELF topological analysis, η(r).25 The ELF study was performed with the TopMod program26 using the corresponding monodeterminantal wavefunctions of the selected structures of the IRC. All computations were carried out with the Gaussian 09 suite of programs.27Conceptual DFT provides different indices to rationalise and understand chemical structure and reactivity.28 The global electrophilicity index,29 ω, is given by the following expression, ω = (μ2/2η), in terms of the electronic chemical potential μ and the chemical hardness η. Both quantities may be approached in terms of the one-electron energies of the frontier molecular orbitals HOMO and LUMO, εH and εL, as μ = (εH + εL)/2 and η = (εL − εH), respectively.30 The global nucleophilicity index,31 N, based on the HOMO energies obtained within the Kohn–Sham scheme,32 is defined as N = EHOMO(Nu) − EHOMO(TCE), where tetracyanoethylene (TCE) is the reference because it presents the lowest HOMO energy in a long series of molecules already investigated in the context of polar organic reactions. This choice allows handling conveniently a nucleophilicity scale of positive values. The electrophilic Pk+ and nucleophilic Pk− Parr functions,33 which allow for the characterisation of the electrophilic and nucleophilic centers of a molecule, were obtained through the analysis of the Mulliken atomic spin density (ASD) of the radical anion of acrolein 16 and the radical cation of nitrone 15, respectively, by single-point energy calculations from the optimised neutral geometries.
Results and discussion
The present theoretical study has been divided into three parts: (i) first, an analysis of the DFT reactivity indices at the ground state of the reagents involved in the 32CA reaction of nitrone 15 with acrolein 16 is carried out; (ii) second, the potential energy surface (PES) associated with this 32CA reaction is explored and characterised; (iii) and finally, a BET study of the endo regioisomeric channels associated with the 32CA reaction between nitrone 15 and acrolein 16 is performed in order to characterise its molecular mechanism.(i) Analysis of the global and local DFT reactivity indices at the ground state of the reagents involved in the 32CA reaction of nitrone 15 with acrolein 16
Studies devoted to DA and 32CA reactions have shown that the analysis of the reactivity indices defined within conceptual DFT28,34 is a powerful tool to understand the reactivity in polar cycloadditions. The global indices, namely, electronic chemical potential μ, chemical hardness η, global electrophilicity ω and global nucleophilicity N, for the reagents involved in this 32CA reaction are given in Table 1.| μ | η | ω | N | |
|---|---|---|---|---|
| DCE 8 | −5.72 | 7.72 | 2.12 | 0.65 |
| Acrolein 16 | −4.58 | 7.78 | 1.34 | 1.76 |
| C-Methyl-N-phenyl nitrone 15 | −3.41 | 6.09 | 0.96 | 3.77 |
| Simplest nitrone 6 | −3.38 | 7.79 | 0.73 | 2.95 |
| Ethylene 11 | −3.38 | 10.05 | 0.57 | 1.83 |
| C,N-Dimethyl nitrone 12 | −2.93 | 7.62 | 0.56 | 3.49 |
| Ynolate 13 | −1.79 | 6.55 | 0.25 | 5.17 |
It can be observed that the electronic chemical potential of nitrone 15, μ = −3.41 eV, is higher than that of acrolein 16, μ = −4.58 eV, indicating that along a polar reaction the GEDT will take place from the nitrone framework towards the acrolein one.
The simplest nitrone 6 presents a nucleophilicity N index of 2.95 eV and an electrophilicity ω index of 0.73 eV, being classified on the borderline of strong nucleophiles,6 and as a moderate electrophile.35 Substitution of the N-hydrogen atom by a methyl group and the incorporation of a phenyl group at the C carbon of nitrone noticeably increases the nucleophilicity N index of nitrone 15 to N = 3.77 eV, and slightly increases its electrophilicity index to ω = 0.96 eV. In consequence, nitrone 15 is considered a strong nucleophile participating in polar reactions.
Polar cycloaddition reactions require the participation of good electrophiles and good nucleophiles. Ethylene 11 is one of the poorest electrophilic, ω = 0.57 eV, and nucleophilic, N = 1.83 eV, species involved in cycloaddition reactions, being classified as a marginal electrophile and a marginal nucleophile. Therefore, it cannot participate in polar reactions.5 Inclusion of a carbonyl group in the ethylene framework notably increases the electrophilicity ω index of acrolein 16, ω = 1.34 eV, being now classified as a strong electrophile, and slightly decreases its nucleophilicity index to N = 1.76 eV, remaining classified as a marginal nucleophile.
Along a polar reaction involving the participation of non-symmetric reagents, the most favourable reactive channel is that involving the initial two-center interaction between the most electrophilic center of the electrophile and the most nucleophilic center of the nucleophile.36 Recently, we have proposed the electrophilic Pk+ and nucleophilic Pk− Parr functions, derived from the changes of the electron density reached via the GEDT process7 from the nucleophile to the electrophile, as powerful tools in the study of the local reactivity in polar processes.33 Analysis of these functions accounts for the most favourable single bond formation between the most electrophilic and nucleophilic centers of the reagents.36 Hence, in order to characterise the most nucleophilic and the most electrophilic centers of the species involved in this 32CA reaction, the nucleophilic Pk− Parr functions of nitrone 15 and the electrophilic Pk+ Parr functions of acrolein 16 were analysed. Fig. 1 shows the 3D representation of the ASD of the radical cation 15+· and the nucleophilic Pk− Parr functions of nitrone 15, and the 3D representation of the ASD of the radical anion 16−· and the electrophilic Pk+ Parr functions of acrolein 16.
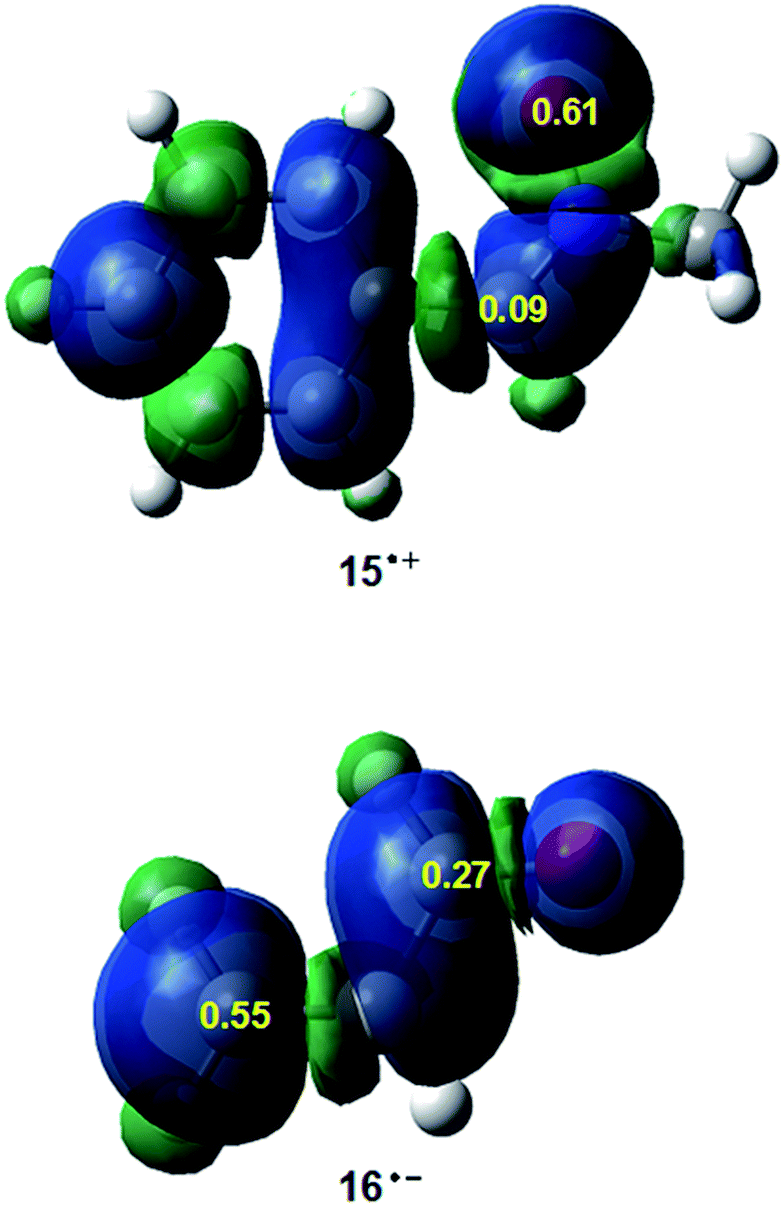 | ||
| Fig. 1 3D representations of the ASD of the radical cation 15+· and the radical anion 16−·, and the nucleophilic Pk− and electrophilic Pk+ Parr functions of nitrone 15 and acrolein 16. | ||
Analysis of the nucleophilic Pk− Parr functions at nitrone 15 indicates that the O1 oxygen is the most nucleophilic center of this species presenting the maximum value, Pk− = 0.61 (see Scheme 5 for atom numbering). At this oxygen, the value of the local nucleophilicity Nk index is NO1 = 2.28 eV.33 Note that the local nucleophilicity Nk index at the C3 carbon in nitrone 15 presents a very low value, NC3 = 0.31 eV. On the other hand, the electrophilic Pk+ Parr functions of acrolein 16 indicates that the terminal C4 carbon is ca. twice as electrophilically activated, Pk+ = 0.55, as the carbonyl C6 carbon, Pk+ = 0.27. The corresponding local electrophilicity ωk values are ωC4 = 0.73 eV and ωC3 = 0.36 eV.33 Therefore, the most favourable electrophile–nucleophile interaction along the nucleophilic attack of nitrone 15 on acrolein 16 will take place between the most nucleophilic center of nitrone 15, the O1 oxygen, and the most electrophilic center of acrolein 16, the C4 carbon.
(ii) Study of the PES of the 32CA reaction of nitrone 15 with acrolein 16
Due to the non-symmetry of the reagents, four reactive channels are feasible along the nucleophilic attack of nitrone 15 on acrolein 16. The two stereoisomeric reactive channels, the endo and the exo ones, are related to the two stereoisomeric approach modes of the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of acrolein 16 relative to the sp2 hybridised N2 nitrogen of nitrone 15. Along the endo channel, the carbonyl group is placed over the nitrogen atom of the nitrone framework. The two regioisomeric reaction pathways are related to the two regioisomeric approach modes of the O1 oxygen or the C3 carbon of nitrone 15 toward the β-conjugated C4 carbon of acrolein; depending on the relative position of the carbonyl group of acrolein 16 with respect to the oxygen O1 atom of nitrone 15 in the isoxazolidines, the two regioisomeric possibilities were named meta and ortho. An exploration of the PES for the zw-type 32CA reaction between nitrone 15 and acrolein 16 indicates that this reaction takes place through a one-step mechanism. Consequently, the reagents, one TS and one cycloadduct were located and characterised along each one of the four reactive channels (see Scheme 5). Relative electronic energies of the stationary points involved in this 32CA reaction, in gas phase and in DCM, are displayed in Table 2, while the total electronic energies are given in Table S1 in the ESI.†
O group of acrolein 16 relative to the sp2 hybridised N2 nitrogen of nitrone 15. Along the endo channel, the carbonyl group is placed over the nitrogen atom of the nitrone framework. The two regioisomeric reaction pathways are related to the two regioisomeric approach modes of the O1 oxygen or the C3 carbon of nitrone 15 toward the β-conjugated C4 carbon of acrolein; depending on the relative position of the carbonyl group of acrolein 16 with respect to the oxygen O1 atom of nitrone 15 in the isoxazolidines, the two regioisomeric possibilities were named meta and ortho. An exploration of the PES for the zw-type 32CA reaction between nitrone 15 and acrolein 16 indicates that this reaction takes place through a one-step mechanism. Consequently, the reagents, one TS and one cycloadduct were located and characterised along each one of the four reactive channels (see Scheme 5). Relative electronic energies of the stationary points involved in this 32CA reaction, in gas phase and in DCM, are displayed in Table 2, while the total electronic energies are given in Table S1 in the ESI.†
| Gas phase | DCM | |
|---|---|---|
| a Relative to nitrone 15 and acrolein 16. | ||
| TS3-mn | 7.9 | 9.0 |
| TS3-mx | 11.9 | 11.1 |
| TS3-on | 10.3 | 11.5 |
| TS3-ox | 13.5 | 13.5 |
| 17-n | −30.4 | −29.1 |
| 17-x | −29.0 | −28.3 |
| 18-n | −32.4 | −29.9 |
| 18-x | −28.5 | −28.5 |
The gas phase activation energies associated with the 32CA reaction between nitrone 15 and acrolein 16 are 7.9 (TS3-mn), 11.9 (TS3-mx), 10.3 (TS3-on) and 13.5 (TS3-ox) kcal mol−1, the reaction being exothermic by between 28 and 30 kcal mol−1. Formation of CA 17-n is kinetically favoured, while that of 18-n is thermodynamically favoured. These energy results indicate that this 32CA reaction is kinetically and moderately regioselective, as the most favourable meta TS3-mn is 2.4 kcal mol−1 below the ortho TS3-on, and displays a complete endo stereoselectivity as the most favourable endo TS3-mn is 4.0 kcal mol−1 below the exo TS3-mx. The strong exothermic character of the reaction makes the formation of the four CAs irreversible; therefore, the major product of the reaction, isoxazolidine 17-n, is formed by kinetic control.
Activation and reaction energies of the endo stereoisomeric channel increase by between 1 and 3 kcal mol−1 (see Table 2) when solvent effects of DCM are considered. This behaviour is a consequence of a better solvation of the reagents than the TSs and CAs.37 In DCM, while the moderate regioselectivity found in gas phase remains unchanged, the complete endo stereoselectivity found in gas phase diminishes; the most favourable endo TS3-mn is now 2.1 kcal mol−1 below the exo TS3-mx.
The geometries of the TSs involved in the zw-type 32CA reaction between nitrone 15 and acrolein 16 are shown in Fig. 2. At the meta TSs, the lengths of the O1–C4 and C3–C5 forming bonds are 1.784 and 2.454 Å at TS3-mn and 1.822 and 2.496 Å at TS3-mx, respectively, while at the ortho TSs the lengths of the O1–C5 and C3–C4 forming bonds are 2.224 and 2.015 Å at TS3-on and 2.183 and 2.066 Å at TS3-ox. These lengths indicate that the TSs involved in the most favourable meta channel are more asynchronous than those involved in the ortho one. The IRCs from the ortho TSs toward [3+2] cycloadducts 18-n and 18-x show that the two ortho stereoisomeric channels are associated to a two-stage one-step mechanism14 initialised by the nucleophilic attack of the O1 oxygen of nitrone 15 on the C4 carbon of acrolein 16.
 | ||
| Fig. 2 MPWB1K/6-31G(d) geometries of the TSs involved in the 32CA reaction of nitrone 15 with acrolein 16. Distances are given in Angstroms. | ||
The electronic nature of the TSs associated with the one-step 32CA reaction of nitrone 15 with acrolein 16 was studied by analysing the bond order (BO)38 and computing the GEDT at the corresponding TSs. In gas phase, the BO values of the O1–C4 and C3–C5 forming bonds at the meta TSs associated with the formation of [3+2] CAs 17-n and 17-x are 0.50 and 0.21 at TS3-mn and 0.47 and 0.21 at TS3-mx, respectively, while the BO values of the O1–C5 and C3–C4 forming bonds at the ortho TSs associated with the formation of CAs 18-n and 18-x are 0.25 and 0.44 at TS3-on and 0.27 and 0.41 at TS3-ox. These values suggest asynchronous bond formation processes, in which the bond formation at the C4 carbon of acrolein 16 is more advanced than that at the C5 carbon atom.
In order to calculate the GEDT at the TSs, the natural atomic charges at the TSs for the meta/ortho regioisomeric channels and the endo/exo stereoisomeric ones, obtained through a natural population analysis (NPA), were shared between the nitrone and the acrolein frameworks. In gas phase, the GEDT that fluxes from the nitrone moiety toward the acrolein one is 0.16e at TS3-mn, 0.14e at TS3-mx, 0.10e at TS3-on and 0.07e at TS3-ox. The most favourable meta/endo channel via TS3-mn is the most polar one, in clear agreement with a zw-type mechanism.3 These low values denote a low polar character of these zw-type 32CA reactions, although suggest a slight acceleration of the 32CA reaction due to the strong nucleophilic and electrophilic character of nitrone 15 and acrolein 16, respectively. Note that the GEDT at the TS associated with the non-polar 32CA reaction between the simplest nitrone 6 and ethylene 11 is 0.01e.5
When comparing the 32CA reaction between nitrone 15 and acrolein 16 with that between the simplest nitrone 6 and DCE 8, the larger acceleration found in the latter 32CA reaction is in agreement with the high GEDT computed at the corresponding TS, 0.26e. In spite of the higher nucleophilic character of C-phenyl nitrone 15 than that of the simplest nitrone 6, the higher electrophilic character of DCE 8 than that of acrolein 16 is responsible for the higher polar character of the 32CA reaction involving ED ethylenes.
Finally, the energy results and geometries obtained by using the MPWB1K functional were compared with those obtained by using the B3LYP one. For this purpose, the stationary points associated with the 32CA reaction between nitrone 15 and acrolein 16 were fully optimised at the B3LYP/6-31G(d) level. Gas phase total and relative energies are given in Table S2,† while the TS geometries are shown in Fig. S1 in the ESI.† A comparison of the gas phase relative energies indicates that while the B3LYP activation energies are found to be between 3.4 and 4.4 kcal mol−1 higher than the MPW1K ones, the reaction energies are underestimated by 17 kcal mol−1. On the other hand, both functionals yield a similar endo stereoselectivity, while the B3LYP functional reduces the moderate meta regioselectivity. A comparison of the B3LYP and MPWB1K lengths of the O–C and C–C forming bonds indicates that both functionals give similar values, thus giving similar bonding changes at the TSs.
(iii) Characterisation of the molecular mechanisms of zw-type 32CA reactions of nitrones with ED ethylenes
Several theoretical studies have shown that the ELF topological analysis of the changes of electron density along a reaction path can be used as a valuable tool to understand the bonding changes along the reaction path, and consequently to establish the molecular mechanisms.12 After an analysis of the electron density, ELF provides basins of attractors, which are the domains in which the probability of finding an electron pair is maximal.39 The spatial points in which the gradient of ELF has a maximum value are designated as attractors. The basins are classified as core basins and valence basins. The latter are characterised by the synaptic order, i.e. the number of atomic valence shells in which they participate. Thus, there are monosynaptic, disynaptic, trisynaptic basins and so on.40 Monosynaptic basins, labelled V(A), correspond to the lone pairs or non-bonding regions, while disynaptic basins, labelled V(A,B), connect the core of two nuclei A and B and, thus, correspond to a bonding region between A and B. This description recovers the Lewis bonding model, providing a very suggestive graphical representation of the molecular system.ELF topological analysis of the structures involved in an elementary step allows characterising mainly three types of valence basins: (i) protonated basins, V(A,H), (ii) monosynaptic basins, V(A), associated with non-bonding regions, and (iii) disynaptic basins, V(A,B), associated with bonding regions. A set of valence basins characterises the ELF topology of a particular molecular structure. Analysis of changes in number or type of valence basins along the IRC allows the characterisation of the IRC points, Pi, defining the different phases that topologically characterise a molecular mechanism.
Recently, Domingo has shown that the formation of the C–C single bond in organic reactions involving sp2 hybridised carbons begins in the short C–C distance range of 1.9–2.0 Å by merging two monosynaptic basins, V(Cx) and V(Cy), into a new disynaptic basin, V(Cx,Cy), associated with the formation of the new Cx–Cy single bond.7 The Cx and Cy carbons characterised by the presence of the monosynaptic basins V(Cx) and V(Cy) are called pseudoradical centers.7
The 32CA reaction between nitrone 15 and acrolein 16 can take place along four competitive channels; two pairs of endo/exo stereoisomeric channels and two pairs of meta/ortho regioisomeric channels. Although the energy differences between the TSs associated with the endo/exo stereoisomeric channels are higher than those associated with the meta/ortho regioisomeric ones, analysis of the geometric and electronic properties of the four TSs indicates that the endo/exo stereoisomeric TSs present similar behaviours, suggesting analogous molecular mechanisms. Consequently, only the regioisomeric reaction paths associated with the most favourable endo regioisomeric channels were selected to perform the BET studies of the 32CA reaction of nitrone 15 with acrolein 16 and, thus, to establish the molecular mechanism of 32CA reactions of nitrones with ED ethylenes.
Finally, an ELF topological analysis of the regioisomeric TSs involved in the 32CA reaction between the simplest nitrone 6 and DCE 8, and the TS involved in the 32CA reaction between dimethylnitrone 12 and ynolate 13, recently studied by Merino et al.,13 was also performed in order to characterise the most significant bonding changes of these TSs.
| MC3-mn | P1 | P2 | P3 | P4 | P5 | P6 | P7 | 17-n | |
|---|---|---|---|---|---|---|---|---|---|
| d(O1–C4) | 2.783 | 2.203 | 1.886 | 1.823 | 1.665 | 1.639 | 1.584 | 1.489 | 1.419 |
| d(C3–C5) | 3.357 | 2.743 | 2.529 | 2.485 | 2.360 | 2.332 | 2.260 | 2.021 | 1.555 |
| GEDT | 0.02 | 0.11 | 0.16 | 0.16 | 0.13 | 0.12 | 0.08 | −0.06 | −0.25 |
| V(O1,N2) | 1.42 | 1.39 | 1.29 | 1.23 | 1.14 | 1.14 | 1.09 | 1.02 | 0.95 |
| V(N2,C3) | 3.88 | 3.82 | 3.95 | 3.04 | 2.64 | 2.60 | 2.51 | 2.23 | 1.86 |
| V(C4,C5) | 1.66 | 3.26 | 3.02 | 2.90 | 2.37 | 2.31 | 2.24 | 2.08 | 1.96 |
| V′(C4,C5) | 1.70 | ||||||||
| V(C5,C6) | 2.34 | 2.32 | 2.47 | 2.44 | 2.57 | 2.56 | 2.50 | 2.31 | 2.07 |
| V(O1) | 3.07 | 2.96 | 2.96 | 2.95 | 2.76 | 2.77 | 2.74 | 2.63 | 2.54 |
| V′(O1) | 2.92 | 2.96 | 2.95 | 2.99 | 3.34 | 2.64 | 2.60 | 2.51 | 2.52 |
| V′′(O1) | 0.71 | ||||||||
| V(O1,C4) | 0.88 | 1.10 | 1.29 | ||||||
| V(C5) | 0.08 | 0.26 | 0.45 | 0.48 | 0.54 | ||||
| V(C3) | 0.18 | 0.21 | 0.30 | ||||||
| V(C3,C5) | 1.24 | 1.84 | |||||||
| V(N2) | 1.02 | 1.45 | 1.51 | 1.64 | 1.96 | 2.31 |
 | ||
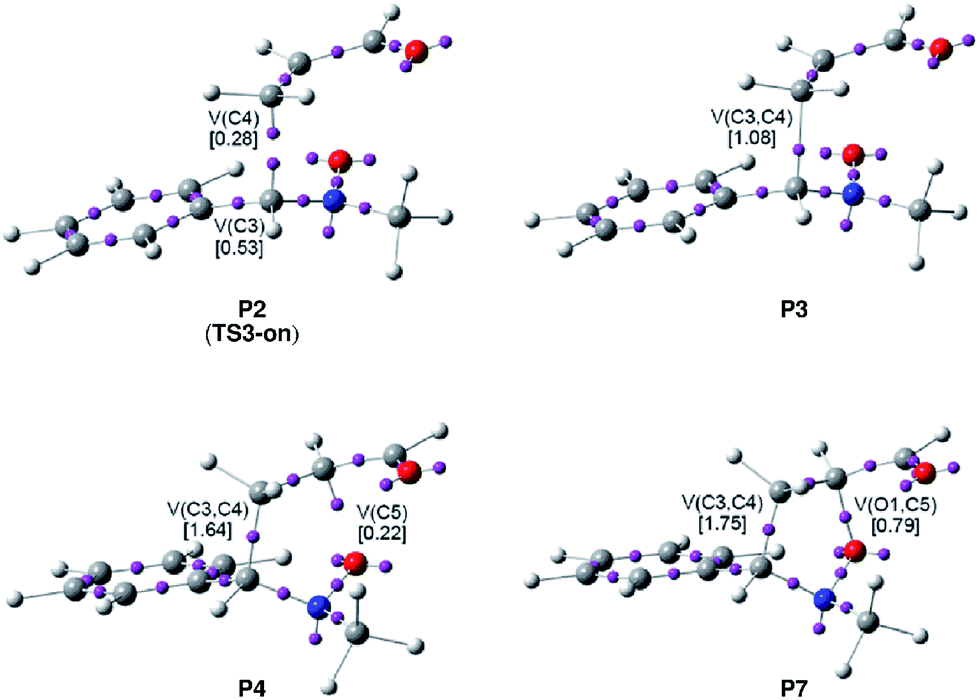
| Fig. 3 ELF attractor positions for the most relevant points of the IRC associated with the O1–C5 and C3–C4 bond formation along the meta/endo regioisomeric channel of the 32CA reaction between nitrone 15 and acrolein 16. | ||
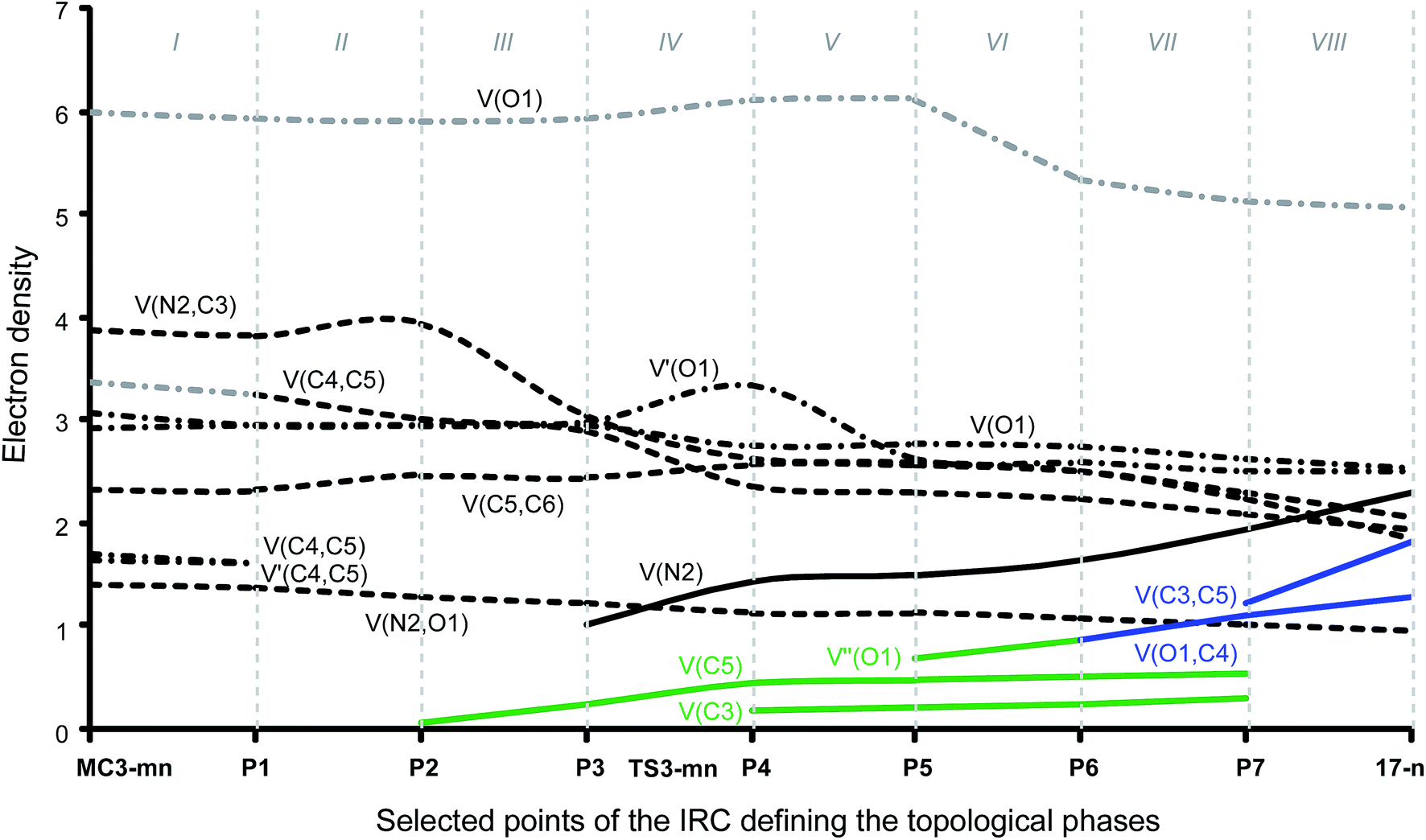 | ||
| Fig. 4 Graphical representation of the basin-population changes along the meta/endo regioisomeric channel of the 32CA reaction between nitrone 15 and acrolein 16. Point-dotted curves in grey represent the sum of disynaptic basins describing a double bond region or monosynaptic basins describing lone pairs. | ||
Phase I, 2.78 Å ≥ d(O1–C4) > 2.20 Å and 3.36 Å ≥ d(C3–C5) > 2.74 Å, begins at MC3-mn, d(O1–C4) = 2.783 Å and d(C3–C5) = 3.357 Å, being a minimum in the PES connecting TS3-mn with the separated reagents 15 and 16. The ELF picture of MC3-mn shows the topological characteristics of the separated reagents. ELF analysis of MC3-mn shows two monosynaptic basins, V(O1) and V′(O1), with a population of 3.07e and 2.92e, associated with the oxygen O1 lone pairs of the nitrone moiety. Two disynaptic basins, V(O1,N2) and V(N2,C3), with a population of 1.42e and 3.88e, which are associated with the O1–N2 single and N2–C3 double bond regions of the nitrone framework, are also observed. Moreover, the ELF topology of MC3-mn shows two disynaptic basins, V(C4,C5) and V′(C4,C5), integrating 1.66e and 1.70e, which belong to the C4–C5 double bond region of the acrolein framework, and one disynaptic basin, V(C5,C6), with a population of 2.34e. At the end of this phase, the GEDT is 0.11e.
Phase II, 2.20 Å ≥ d(O1–C4) > 1.89 Å and 2.74 Å ≥ d(C3–C5) > 2.53 Å, starts at P1. At this point, the two V(C4,C5) and V′(C4,C5) disynaptic basins present at MC3-mn have merged into a new V(C4,C5) disynaptic basin, integrating 3.26e. This topological change is the consequence of the loss of electron density in the C4–C5 bonding region, ca. 0.10e.
Phase III, 1.89 Å ≥ d(O1–C4) > 1.82 Å and 2.53 Å ≥ d(C3–C5) > 2.49 Å, begins at P2. The most significant occurrence in this phase is the creation of a new V(C5) monosynaptic basin at P2, integrating 0.08e (see P2 in Fig. 4). The electron density of the new V(C5) monosynaptic basin is mainly reached through the depopulation of the V(C4,C5) disynaptic basin, which continues its depopulation until reaching a population of 3.02e. This loss of electron density is mainly gathered at the V(C5,C6) disynaptic basin, which has increased its integration, 2.47e. At P2 the maximum GEDT along the 32CA reaction is observed, 0.16e, indicating that this zw-type 32CA has a low polar nature.
Phase IV, 1.82 Å ≥ d(O1–C4) > 1.67 Å and 2.49 Å ≥ d(C3–C5) > 2.36 Å, begins at P3, where, together with the depopulation of the V(O1,N2) and V(N2,C3) disynaptic basins by 0.06e and 0.91e, a new V(N2) monosynaptic basin, integrating 1.02e, is created at the N2 nitrogen atom. This monosynaptic basin, whose electron density proceeds mainly from the depopulation of the V(N2,C3) disynaptic basin, is associated with the nitrogen N2 lone pair present in isoxazolidine 17-n. At this phase, the TS of the reaction, TS3-mn, d(O1–C4) = 1.784 Å and d(C3–C5) = 2.454 Å, is found. At TS3-mn only slight changes in electron density with respect to those found at P3 are observed (see Tables 3 and 5). The GEDT at TS3-mn is 0.16e.
At phase V, 1.67 Å ≥ d(O1–C4) > 1.64 Å and 2.36 Å ≥ d(C3–C5) > 2.33 Å, the most notable change is the creation of a new V(C3) monosynaptic basin at P4, integrating 0.18e, whereas the V(C5) monosynaptic basin created in phase III has reached a population of 0.45e, as a consequence of the strong depopulation experienced by the V(C4,C5) disynaptic basin, 0.53e. Note that the two V(C3) and V(C5) monosynaptic basins are required for the formation of the C3–C5 single bond in phase VII. Meanwhile, the V(N2) monosynaptic basin has increased its population to 1.45e as the consequence of the depopulation of both the V(O1,N2) and V(N2,C3) disynaptic basins. GEDT has slightly decreased to 0.13e.
Phase VI, 1.64 Å ≥ d(O1–C4) > 1.58 Å and 2.33 Å ≥ d(C3–C5) > 2.26 Å, begins at P5. The most relevant change along this short phase is the remarkable creation of a V′′(O1) monosynaptic basin integrating 0.71e as a result of the decrease of the electron density of the V′(O1) monosynaptic basin from 3.34e to 2.64e. GEDT scarcely varies.
Phase VII, 1.58 Å ≥ d(O1–C4) > 1.49 Å and 2.26 Å ≥ d(C3–C5) > 2.02 Å, begins at P6. The first most relevant change along the reaction path takes place at this point. While the V′′(O1) monosynaptic basin created in the previous phase has disappeared, a new V(O1,C4) disynaptic basin is created with an initial population of 0.88e (see the V(O1,C4) disynaptic basin in P6 in Fig. 3, and the change from V′′(O1), in green in P5, to V(O1,C4), in blue in P6, in Fig. 4). This notable topological change indicates that the formation of the new O1–C4 single bond has already begun at a length of ca. 1.58 Å. GEDT decreases to 0.08e.
Finally, phase VIII, 1.49 Å ≥ d(O1–C4) ≥ 1.42 Å and 2.02 Å ≥ d(C3–C5) ≥ 1.56 Å, begins at P7 and ends at isoxazolidine 17-n, d(O1–C4) = 1.419 Å and d(C3–C5) = 1.555 Å. The second most relevant change along the reaction path takes place at P7. The two V(C3) and V(C5) monosynaptic basins present in the previous phase are merged into a new V(C3,C4) disynaptic basin, which presents a population of 1.24e (see the V(C3,C5) disynaptic basin in P7 in Fig. 3, and the merger of V(C3) and V(C5), in green in P6, into the new V(C3,C5), in blue in P7, in Fig. 4). This notable topological change indicates that the formation of the new C3–C5 single bond has already begun at a length of ca. 2.02 Å, by a C-to-C coupling of the two C3 and C5 pseudoradical centers. With the start of the C3–C5 bond formation, GEDT decreases until reaching a slightly negative value, −0.06e, as the consequence of a back donation process. From P7 to 17-n only changes in basin populations are observed. Some topological features in cycloadduct 17-n are noteworthy: the O1–C4 and O1–N2 single bond regions are very polarised toward the oxygen atom, the population of the V(O1,C4) and V(O1,N2) disynaptic basins and that of the V(O1) and V′(O1) monosynaptic basins integrating 1.29e and 0.95e, and 2.54e and 2.52e, respectively. The nitrogen N2 lone pair and the V(C3,C5) disynaptic basin have increased their populations to 2.31e and 1.84e. Interestingly, GEDT at 17-n has a large negative value, −0.25e, due to the strong polarisation of the O1–C4 single bond.
| MC3-on | P1 | P2 | P3 | P4 | P5 | P6 | P7 | 18-n | |
|---|---|---|---|---|---|---|---|---|---|
| d(C3–C4) | 3.408 | 2.121 | 2.015 | 1.911 | 1.639 | 1.600 | 1.579 | 1.569 | 1.522 |
| d(O1–C5) | 2.844 | 2.277 | 2.224 | 2.168 | 1.906 | 1.790 | 1.694 | 1.634 | 1.409 |
| GEDT | 0.03 | 0.11 | 0.10 | 0.09 | 0.00 | −0.13 | −0.18 | −0.20 | −0.32 |
| V(O1,N2) | 1.42 | 1.35 | 1.31 | 1.25 | 1.11 | 1.08 | 1.03 | 1.00 | 0.90 |
| V(N2,C3) | 3.83 | 2.72 | 2.48 | 2.30 | 1.95 | 1.90 | 1.90 | 1.89 | 1.85 |
| V(C4,C5) | 1.67 | 3.19 | 2.87 | 2.64 | 2.19 | 2.11 | 2.05 | 2.04 | 1.90 |
| V′(C4,C5) | 1.69 | ||||||||
| V(C5,C6) | 2.21 | 2.35 | 2.38 | 2.46 | 2.33 | 2.28 | 2.21 | 2.17 | 2.12 |
| V(O1) | 2.94 | 2.86 | 2.85 | 2.89 | 2.95 | 3.08 | 3.43 | 2.77 | 2.61 |
| V′(O1) | 3.05 | 2.94 | 2.90 | 2.89 | 2.84 | 2.82 | 2.77 | 2.73 | 2.48 |
| V(N2) | 0.96 | 1.21 | 1.42 | 1.95 | 2.10 | 2.16 | 2.17 | 2.37 | |
| V(C3) | 0.39 | 0.53 | |||||||
| V(C4) | 0.28 | ||||||||
| V(C3,C4) | 1.08 | 1.64 | 1.71 | 1.75 | 1.77 | 1.85 | |||
| V(C5) | 0.22 | 0.16 | |||||||
| V(O1,C5) | 0.79 | 1.32 |
 | ||
| Fig. 5 ELF attractor positions for the most relevant points of the IRC associated with the formation of the C3–C4 and O1–C5 single bonds along the ortho/endo regioisomeric channel of the 32CA reaction between nitrone 15 and acrolein 16. | ||
 | ||
| Fig. 6 Graphical representation of the basin-population changes along the ortho/endo regioisomeric channel of the 32CA reaction between nitrone 15 and acrolein 16. Point-dotted curves in grey represent the sum of disynaptic basins describing a double bond region or monosynaptic basins describing lone pairs. | ||
Phase I, 3.41 Å ≥ d(C3–C4) > 2.12 Å and 2.84 Å ≥ d(O1–C5) > 2.28 Å, begins at MC3-on, d(C3–C4) = 3.408 Å and d(O1–C5) = 2.844 Å, which is a minimum in the PES connecting the separated reagents 15 and 16 with the corresponding TS3-on. Just as MC3-mn, the ELF picture of MC3-on mainly recovers the whole topological behaviour of the separated reagents. ELF analysis of MC3-on shows the presence of two monosynaptic basins, V(O1) and V′(O1), with populations of 2.94e and 3.05e, associated with the oxygen O1 lone pairs of the nitrone moiety. Two disynaptic basins, V(O1,N2) and V(N2,C3), associated with the O1–N2 single and N2–C3 double bond regions of the nitrone moiety are also observed with populations of 1.42e and 3.83e. On the other hand, ELF topology of MC3-on shows the presence of two disynaptic basins, V(C4,C5) and V′(C4,C5), integrating 1.67e and 1.69e, belonging to the C4–C5 double bond region of the acrolein moiety, and one disynaptic basin, V(C5,C6), with a population of 2.21e.
Phase II, 2.12 Å ≥ d(C3–C4) > 2.02 Å and 2.27 Å ≥ d(O1–C5) > 2.22 Å, begins at P1. At this point the most relevant change is the formation of a new V(C3) monosynaptic basin at the nitrone fragment, which integrates 0.39e, together with the depopulation of the V(N2,C3) and V(O1,N2) disynaptic basins to 2.72e and 1.35e. In this phase, a new V(N2) monosynaptic basin appears with a population of 0.96e. This monosynaptic basin is associated with the nitrogen N2 lone pair present at isoxazolidine 18-n. It is also observed that the two V(C4,C5) and V′(C4,C5) disynaptic basins belonging to the C4–C5 double bond region of the acrolein framework have merged into one V(C4,C5) disynaptic basin integrating 3.19e.
The short Phase III, 2.02 Å ≥ d(C3–C4) > 1.91 Å and 2.22 Å ≥ d(O1–C5) > 2.17 Å, begins at P2, d(C3–C4) = 2.015 Å and d(O1–C5) = 2.224 Å. Structure P2 corresponds to the TS of the reaction, TS3-on. The most significant change occurring at this phase is the creation of a new V(C4) monosynaptic basin at the acrolein moiety, integrating 0.28e. Note that the C4 carbon of acrolein is the most electrophilic center of this molecule. On the other hand, the populations of the V(C3) and V(N2) monosynaptic basins increase to 0.53e and 1.21e.
Phase IV, 1.91 Å ≥ d(C3–C4) > 1.64 Å and 2.17 Å ≥ d(O1–C5) > 1.91 Å, begins at P3. A second significant event along the reaction path occurs in this phase. While the two V(C3) and V(C4) monosynaptic basins disappear, a new V(C3,C4) disynaptic basin, integrating 1.08e, is created (see the V(C3,C4) disynaptic basin in P3 in Fig. 5, and the merger of V(C3) and V(C4), in green in TS3-on, into the new V(C3,C4), in blue in P3, in Fig. 6). This notable topological change indicates that the formation of the new C3–C4 single bond has already begun at a length of ca. 1.91 Å, by a C-to-C coupling of the two C3 and C4 pseudoradical centers. On the other hand, the population of the V(N2) monosynaptic basin increases to 1.42e along the reaction progress, whereas the population of V(C4,C5) decreases to 2.64e.
Phase V, 1.64 Å ≥ d(C3–C4) > 1.60 Å and 1.91 Å ≥ d(O1–C5) > 1.79 Å, begins at P4. At this point, a new V(C5) monosynaptic basin is created at the C5 carbon integrating 0.22e. At this phase, the population of the recently created V(C3,C4) disynaptic basin and that of the V(N2) monosynaptic basin increases to 1.64e and 1.95e, whereas the populations of the V(O1,N2) and V(N2,C3) disynaptic basins decrease to 1.11e and 1.95e, respectively, as the reaction progresses.
Phase VI, 1.60 Å ≥ d(C3–C4) > 1.58 Å and 1.79 Å ≥ d(O1–C5) > 1.69 Å, begins at P5. At this point, there is a slight decrease in the population of the V(C5) monosynaptic basin to 0.16e, as well as in the populations associated with the V(O1,N2), V(N2,C3) and V(C4,C5) disynaptic basins, i.e. 1.08e, 1.90e and 2.11e. On the other hand, the populations of the V(N2) monosynaptic and V(C3,C4) disynaptic basins increase to 2.10e and 1.71e, respectively.
Phase VII, 1.58 Å ≥ d(C3–C4) > 1.57 Å and 1.69 Å ≥ d(O1–C5) > 1.63 Å, begins at P6. At this point, while the populations of the two V(O1) and V′(O1) monosynaptic basins are strongly increased to reach a total population of 6.20e, the V(C5) monosynaptic basin created in phase V has disappeared.
Phase VIII, 1.57 Å ≥ d(C3–C4) ≥ 1.52 Å and 1.63 Å ≥ d(O1–C5) ≥ 1.40 Å, begins at P7 and ends at 18-n. Another relevant change along the reaction path takes place at P8. Whereas the population of the V(O1) monosynaptic basins decreases to 2.73e, a new V(O1,C5) disynaptic basin is created with an initial integration of 0.79e (see the V(O1,C5) disynaptic basin in P7 in Fig. 5, and the creation of V(O1,C5) in blue in P7 in Fig. 6). This notable topological change indicates that the formation of the new O1–C5 single bond has already begun at a length of ca. 1.63 Å. Along this last phase, the population of the created V(O1,C5) disynaptic basin is increased until reaching 1.32e in 18-n, while the populations of the V(O1) and V′(O1) monosynaptic basins decrease to 2.61e and 2.48e, respectively. These values indicate that the created O1–C5 single bond is strongly polarised toward the O1 oxygen atom. In 18-n, the C3–C4 single bond has reached a population of 1.85e.
Some appealing conclusions can be drawn from the BET study of the two endo regioisomeric channels:
(i) the BET study of the two endo regioisomeric reaction channels allows distinguishing eight differentiated phases associated with the creation or disappearance of valence basins;
(ii) formation of the C–C single bond follows the recently proposed pattern:7 (a) depopulation of the double bond regions; (b) formation of two non-bonding V(C) and V(C′) monosynaptic basins; and (c) formation of a new V(C,C′) disynaptic basin through the merger of the electron density of the aforementioned monosynaptic basins. The formation of the C–C single bond begins at 2.02 Å along the meta channel and at 2.17 Å along the ortho one;
(iii) a different behaviour is found for the formation of the O–C single bond. Along the most favourable meta channel, the formation of the O1–C4 single bond begins with the creation of a new V(O1,C4) disynaptic basin at the very short distance of 1.58 Å. Interestingly, the electron population of this new disynaptic basin mainly proceeds from the electron density of one V(O1) monosynaptic basin associated with the lone pairs of the nitrone O1 oxygen atom. Likewise, along the ortho regioisomeric channel, the formation of the O1–C5 single bond begins with the creation of a new V(O1,C5) disynaptic basin at the very short distance of 1.63 Å. The electron density of this new V(O1,C5) disynaptic basin proceeds mainly from the electron density of one V(O1) monosynaptic basin associated with the lone pairs of the nitrone O1 oxygen atom. This pattern for the formation of these O–C single bonds is similar to that recently found for the formation of the N–C single bond along ketene–imine Staudinger reactions, in which the electron density of the new N–C single bond comes from the nitrogen lone pair;41
(iv) interestingly, while the formation of the C–C single bond begins at ca. 2.0 Å, the formation of the O–C single bond begins at the very short distance of 1.6 Å. These behaviours indicate that in most 32CA reactions the asynchronicity of the C–C and O–C single bond formation cannot be measured by using geometrical parameters. Thus, while the analysis of the bond lengths of the two O–C and C–C forming bonds suggests that TS3-mn is more asynchronous than TS3-on, analysis of the basin-population changes in Fig. 4 and 6 indicates that the O–C and C–C bond formation is more synchronous along the most favourable meta regioisomeric channel. Similarly, the BO values of the O1–C4 and C3–C5 forming bonds at TS3-mn and those of the O1–C5 and C3–C4 forming bonds at TS3-on suggest an asynchronous bond formation process, in which the bond formation at the C4 carbon of acrolein 16 is more advanced than that at the C5 carbon atom. However, the ELF topological analysis at TS3-mn indicates that the formation of both single bonds is almost synchronous. Moreover, the BO value of the O1–C4 forming bond at TS3-mn, 0.50, and that of the C3–C4 forming bond at TS3-on, 0.44, show that they have high values, but the ELF topological analysis at the corresponding TSs indicates that the formation of these single bonds has not begun. Consequently, while the BO values in the range from 1.0 to 3.0 can be used as a measure of the bonding electron density between two neighbouring atoms, BO values in the range from 0.0 to 1.0 cannot account for any information about the bonding evolution along the reaction coordinate.
(v) along the two endo regioisomeric channels, the formation of the O1–C4 or C3–C4 single bonds begins by a two-center interaction involving the most electrophilic center of acrolein 16, the C4 carbon. Note that at the most favourable meta/endo channel, the O1–C4 two-center interaction also involves the most nucleophilic center of nitrone 15, the O1 oxygen, a behaviour anticipated by the analysis of the electrophilic and nucleophilic Parr functions;
(vi) along the ortho/endo regioisomeric channel, formation of the second O1–C5 single bond begins at the seventh phase, in which the first C3–C4 single bond is practically formed. This behaviour indicates that the ortho reaction takes place through a two-stage one-step mechanism;14 along the most favourable meta/endo regioisomeric channel, formation of the O1–C4 and C3–C5 single bonds is more synchronous.
| TS1-m | TS3-mn | TS1-o | TS3-on | TS2-o | ||
|---|---|---|---|---|---|---|
| d(O1–C4) | 1.766 | 1.784 | d(O1–C5) | 2.312 | 2.224 | 3.231 |
| d(C3–C5) | 2.522 | 2.454 | d(C3–C4) | 2.074 | 2.015 | 2.123 |
| GEDT | 0.26 | 0.16 | 0.19 | 0.10 | −0.30 | |
| V(O1,N2) | 1.23 | 1.20 | V(O1,N2) | 1.37 | 1.31 | 1.12 |
| V(N2,C3) | 3.85 | 2.96 | V(N2,C3) | 3.41 | 2.48 | 2.88 |
| V(C4,C5) | 2.83 | 2.76 | V(C4,C5) | 3.06 | 2.87 | 2.45 |
| V′(C4,C5) | V′(C4,C5) | 2.41 | ||||
| V(C5,C6) | 2.31 | 2.46 | V(C5,C6) | 2.34 | 2.38 | 2.07 |
| V(O1) | 3.07 | 2.93 | V(O1) | 2.90 | 2.85 | 3.33 |
| V′(O1) | 2.88 | 3.05 | V′(O1) | 2.84 | 2.90 | 2.98 |
| V(C3) | V(C3) | 0.48 | 0.53 | |||
| V(C5) | 0.45 | 0.30 | V(C4) | 0.22 | 0.28 | 0.89 |
| V(N2) | 1.17 | V(N2) | 1.21 | 1.44 |
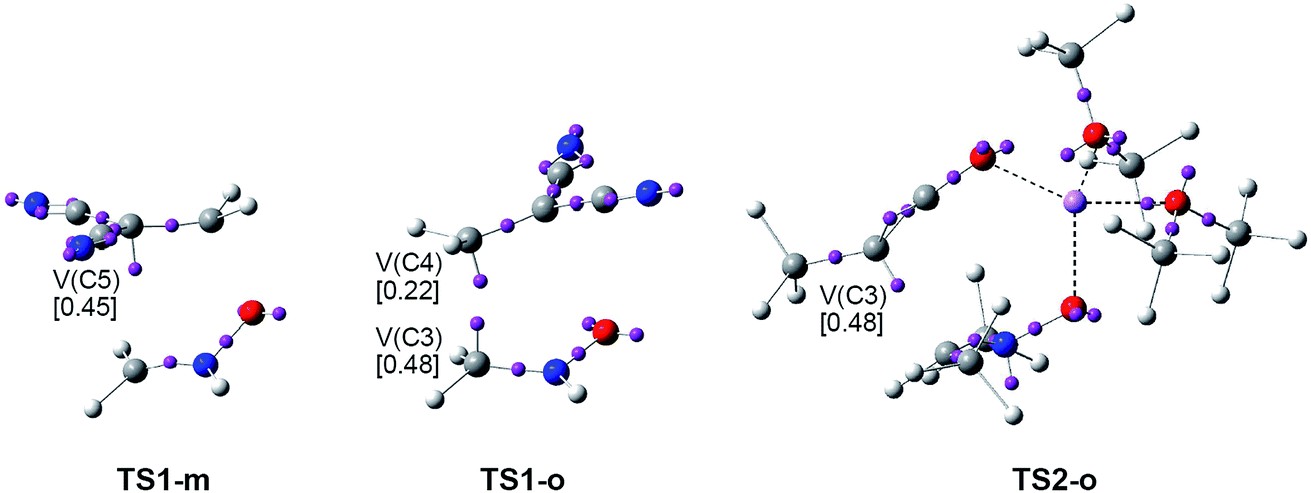 | ||
| Fig. 7 ELF attractor positions for the regioisomeric TSs involved in the 32CA reaction between the simplest nitrone 6 and DCE 8, and the TS involved in the 32CA reaction between dimethylnitrone 12 and ynolate 13. | ||
The ELF picture of the most favourable regioisomeric meta TS involved in the 32CA between the simplest nitrone 6 and DCE 8, TS1-m, d(O1–C4) = 1.766 Å and d(C3–C5) = 2.522 Å, shows the presence of two V(O1,N2) and V(N2,C3) disynaptic basins with populations of 1.23e and 3.85e, belonging to the O1–N2 and N2–C3 bonding regions of the nitrone framework, as well as one V(C4,C5) disynaptic basin integrating 2.85e, belonging to the C4–C5 bonding region of DCE 8. ELF topology of TS1-m also shows the presence of one V(C5) monosynaptic basin integrating 0.45e.
Similarly, the ELF picture of the ortho TS, TS1-o, d(C3–C4) = 2.074 Å and d(O1–C5) = 2.312 Å, shows the presence of two V(O1,N2) and V(N2,C3) disynaptic basins, with populations of 1.37e and 3.41e, belonging to the O1–N2 and N2–C3 bonding regions of the nitrone moiety, as well as one V(C4,C5) disynaptic basin, integrating 3.06e, associated with the C4–C5 bonding region of DCE 8. The most relevant feature of this TS is the presence of two V(C3) and V(C4) monosynaptic basins integrating 0.48e and 0.22e, which are demanded for the subsequent formation of the C3–C4 single bond.
Otherwise, the ELF picture of the TS involved in the 32CA reaction between dimethylnitrone 12 and ynolate 13, TS2-o, d(C3–C4) = 2.123 Å and d(O1–C5) = 3.231 Å, shows that the O1–N2 and N2–C3 bonding regions remain characterised by two V(O1,N2) and V(N2,C3) disynaptic basins whose populations are 1.12e and 2.88e, while the C4–C5 triple bond region of ynolate 13 is characterised by the presence of two V(C4,C5) and V′(C4,C5) disynaptic basins, which integrate a total of 4.86e. The most relevant feature of this TS is the presence of one V(C4) monosynaptic basin, integrating 0.89e, and one V(N2) monosynaptic basin, with a population of 1.44e, which is related to the N2 nitrogen lone pair present in CA 14. While the electron population of the V(C4) monosynaptic basin at this TS comes from the depopulation of the C4–C5 triple bond region of ynolate 13, the V(N2) monosynaptic basin comes from the depopulation of the N2–C3 double bond region of nitrone 12.
Some significant conclusions can be drawn from the ELF topological analysis of the TSs involved in the 32CA reactions of nitrones 6, 12 and 15 towards ED ethylenes 8 and 16 and electron-rich ethylene 13:
(i) ELF pictures of the most favourable meta TS1-m and TS3-mn show the presence of a V(C5) monosynaptic basin with a similar population. No V(C4) monosynaptic basin is observed at both TSs;
(ii) ELF pictures of ortho TS1-o and TS3-on show the presence of two V(C3) and V(C4) monosynaptic basins, with similar populations. Note that these monosynaptic basins are involved in the subsequent formation of the C3–C4 single bond.
(iii) unlike TS3-mn and TS3-on, no V(N2) monosynaptic basin is observed at TS1-m nor at TS1-o. This behaviour, together with the larger population of the V(N2,C3) disynaptic basin at the latter TS1-m and TS1-o, indicates that the changes in the N2–C3 bonding region are more delayed at the simplest nitrone 6;
(iv) in spite of these topological differences, the two regioisomeric TSs involved in the 32CA reaction of the simplest nitrone 6 with DCE 8 show a great similarity along the formation of the O–C and C–C single bonds with that observed in the 32CA reaction of nitrone 15 with acrolein 16. This finding supports a similar description for the formation of the O–C and C–C single bonds along the two regioisomeric channels of 32CA reactions of nitrones with ED ethylenes;
(v) a comparison of the ELF features of ortho TS2-o with those of TS1-o and TS3-on shows significant dissimilarities. Thus, TS2-o shows the presence of a V(C4) monosynaptic basin with high electron population, 0.89e. This behaviour could be due to a notable difference between these 32CA reactions. Analysis of the electronic chemical potential of dimethyl nitrone 12 and ynolate 13, −2.93 and −1.79 eV (see Table 1), indicates that along a polar reaction the GEDT will flux from ynolate 13 toward nitrone 12 (see the change of the sign of the GEDT at TS2-o in Table 5). Consequently, in this zw-type 32CA reaction nitrone 12 will be the electrophile, while ynolate 13 will be the nucleophile. Interestingly, in spite of the high GEDT found at TS2-o, 0.30e, the activation energy associated with this polar process is very high, 14.7 kcal mol−1, as a consequence of the marginal electrophilic character of nitrone 12, 0.56 eV (see Table 1).
In 1965, Woodward and Hoffmann established the pericyclic reactions42 as “reactions in which all first order changes in bonding relationship take place in concert on a close curve”.43 For 32CA reactions, the pericyclic model assumes that the six electrons involved in the cyclisation, four of the TAC and two of the alkene, move in a concerted fashion around the five atoms. BET analysis of the zw-type 32CA reaction between nitrone 15 with acrolein 16 permits to establish that bonding changes are not neither concerted, but sequential along the nucleophilic attack of the nitrone 15 to the ED ethylene 16, nor on a close curve, but the electron density at the beginning of the reaction fluxes from the nucleophilic nitrone to the ED ethylene. In addition, any bonding disynaptic basins in the C–C and O–C forming bond regions exist at TS1-m, TS1-o, TS2-o, TS3-mn and TS3-on, such as it would be expected at the TSs associated with a pericyclic reaction. Consequently, as in Diels–Alder reactions, the BET study of these zw-type 32CA reactions allows rejecting the pericyclic mechanism proposed for them.44
Conclusions
The mechanisms of zw-type 32CA reactions of nitrones with ED ethylenes have been studied using DFT methods at the MPWB1K/6-31G(d) level, and the obtained results are supported by the combination of the analysis of the values of reactivity indices at the ground state of the reagents, derived from conceptual DFT, the exploration and characterisation of the PESs associated with the cycloaddition reaction of nitrone 15 with acrolein 16, and the BET study of the two endo regioisomeric channels.Nitrones such as 15 present a high nucleophilicity N index, being classified as strong nucleophiles. Consequently, it is expected that they participate in zw-type 32CA reactions towards ED ethylenes such as acrolein 16 or DCE 8. In addition, analysis of the nucleophilic Pk− Parr functions allows characterising the O1 oxygen atom as the most nucleophilic center of nitrone 15.
An exploration of the PESs associated with the four competitive reactive channels of the 32CA reaction of nitrone 15 with acrolein 16 indicates that the cycloaddition reaction takes place through a one-step mechanism. This cycloaddition reaction presents moderate meta regioselectivity and complete endo stereoselectivity. In DCM, the most favourable meta/endo reactive channel presents an activation energy of 9.0 kcal mol−1, while the formation of the corresponding cycloadduct is exothermic by 29.1 kcal mol−1.
A BET study of the endo regioisomeric channels allows establishing the molecular mechanism of these relevant 32CA reactions involving nitrones and ED ethylenes, and provides new insight into the bonding changes taking place along the two regioisomeric reaction channels. While the formation of the C–C single bond begins at ca. 2.0 Å, the formation of the O–C bond begins at the very short distance of 1.6 Å. Unlike the formation of the C–C bond, which follows Domingo's recently proposed model,7 the formation of the O–C bond takes place at the short distance of 1.6 Å through the donation of some electron density of the nitrone oxygen lone pairs to the acceptor carbon atom of the ED ethylene. This model for the formation of heteroatom-carbon single bonds is similar to that found in ketene–imine Staudinger reactions.41
A comparative ELF topological analysis of the TSs involved in the 32CA reaction between the simplest nitrone 6 and ED ethylene 8 with those involved in the 32CA reaction between nitrone 15 and acrolein 16 shows a similar mechanism for the formation of the O–C and C–C single bonds along the two regioisomeric channels. This similarity allows us to propose the present BET study as a general model for the bonding changes along the two regioisomeric channels associated with 32CA reactions of nucleophilic nitrones with ED ethylenes.
The present BET study of the one-step mechanisms associated with the two regioisomeric channels of these zw-type 32CA reactions makes it possible to reject the pericyclic mechanism proposed for these cycloaddition reactions.44
Acknowledgements
This work has been supported by the Ministerio de Economía y Competitividad of the Spanish Government, project CTQ2013-45646-P, by FONDECYT through Project 1140341, by the Millennium Nucleus of Chemical Processes and Catalysis (CPC), grant number NC 120082 and by DI-UNAB-793-15/R. M. R.-G. thanks the Ministerio de Economía y Competitividad for a pre-doctoral contract co-financed by the European Social Fund (BES-2014-068258).References
- (a) K. V. Gothelf and K. A. Jørgensen, Chem. Rev., 1999, 82, 327–350 CAS; (b) S. Karlsson and H. E. Hogberg, Org. Prep. Proced. Int., 2001, 33, 105–172 CrossRef PubMed; (c) W. S. Jen, J. J. M. Wiener and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 9874–9875 CrossRef CAS; (d) K. V. Gothelf and K. A. Jørgensen, Chem. Commun., 2000, 1449–1458 RSC.
- A. Piperno, S. V. Giofre, D. Iannazzo, R. Romeo, G. Romeo, U. Chiacchio, A. Rescifina and D. G. Piotrowska, J. Org. Chem., 2010, 75, 2798–2805 CrossRef CAS PubMed.
- L. R. Domingo and S. R. Emamian, Tetrahedron, 2014, 70, 1267–1273 CrossRef CAS PubMed.
- A. Padwa and W. H. Pearson, Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, John Wiley & Sons, Hoboken, New Jersey, 2002, vol. 59 Search PubMed.
- L. R. Domingo, M. J. Aurell and P. Pérez, Tetrahedron, 2014, 70, 4519–4525 CrossRef CAS PubMed.
- P. Jaramillo, L. R. Domingo, E. Chamorro and P. Pérez, J. Mol. Struct.: THEOCHEM, 2008, 865, 68–72 CrossRef CAS PubMed.
- L. R. Domingo, RSC Adv., 2014, 4, 32415–32428 RSC.
- (a) V. Polo, J. Andrés, S. Berski, L. R. Domingo and B. Silvi, J. Phys. Chem. A, 2008, 112, 7128–7134 CrossRef CAS PubMed; (b) J. Andrés, P. González-Navarrete and V. S. Safont, Int. J. Quantum Chem., 2014, 114, 1239–1252 CrossRef PubMed; (c) J. Andrés, S. Berski, L. R. Domingo, V. Polo and B. Silvi, Curr. Org. Chem., 2011, 15, 3566–3575 CrossRef; (d) J. Andrés, P. González-Navarrete and V. S. Safont, Int. J. Quantum Chem., 2014, 114, 1239–1252 CrossRef PubMed; (e) J. Andrés, L. Gracia, P. González-Navarrete and V. S. Safont, Comput. Theor. Chem., 2015, 1053, 17–30 CrossRef PubMed; (f) J. Andrés, L. Gracia, P. González-Navarrete and V. S. Safont, Quantum Chemical Topology Approach for Dissecting Chemical Structure and Reactivity in Applications of Topological Methods in Molecular Chemistry, Springer book series: Challenges and Advances in Computational Chemistry and Physics, ed. E. Alikhani, R. Chauvin, C. Lepetit and B. Silvi, Springer, 2015 Search PubMed.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS PubMed.
- (a) R. Thom, Structural Stability and Morphogenesis: An Outline of a General Theory of Models, Inc.:Reading, MA, 1976 Search PubMed; (b) A. E. R. Woodcock and T. Poston, A Geometrical Study of Elementary Catastrophes, Springer-Verlag, Berlin, 1974 Search PubMed; (c) R. Gilmore, Catastrophe Theory for Scientists and Engineers, Dover, New York, 1981 Search PubMed.
- X. Krokidis, S. Noury and B. Silvi, J. Phys. Chem. A, 1997, 101, 7277–7282 CrossRef CAS.
- (a) S. Berski, J. Andrés, B. Silvi and L. R. Domingo, J. Phys. Chem. A, 2003, 107, 6014–6024 CrossRef CAS; (b) S. Berski, J. Andrés, B. Silvi and L. R. Domingo, J. Phys. Chem. A, 2006, 110, 13939–13947 CrossRef CAS PubMed; (c) I. Viciano, P. González-Navarrete, J. Andrés and S. Martí, J. Chem. Theory Comput., 2015, 11, 1470–1480 CrossRef CAS.
- D. Roca-Lopez, V. Polo, T. Tejero and P. Merino, J. Org. Chem., 2015, 80, 4076–4083 CrossRef CAS PubMed.
- L. R. Domingo, J. A. Saéz, R. J. Zaragozá and M. Arnó, J. Org. Chem., 2008, 73, 8791–8799 CrossRef CAS PubMed.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- (a) C. E. Check and T. M. Gilbert, J. Org. Chem., 2005, 70, 9828–9834 CrossRef CAS PubMed; (b) G. O. Jones, V. A. Guner and K. N. Houk, J. Phys. Chem. A, 2006, 110, 1216–1224 CrossRef CAS PubMed; (c) G. A. Griffith, I. H. Hillier, A. C. Moralee, J. M. Percy, R. Roig and M. K. Vicent, J. Am. Chem. Soc., 2006, 128, 13130–13141 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2004, 108, 6908–6918 CrossRef CAS.
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- (a) H. B. Schlegel, J. Comput. Chem., 1982, 3, 214–218 CrossRef CAS PubMed; (b) Modern Electronic Structure Theory, ed. H. B. Schlegel and D. R. Yarkony, World Scientific Publishing, Singapore, 1994 Search PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
- (a) C. González and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5525 CrossRef; (b) C. González and H. B. Schlegel, J. Chem. Phys., 1991, 95, 5853–5860 CrossRef PubMed.
- (a) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS; (b) B. Y. Simkin and I. Sheikhet, Quantum Chemical and Statistical Theory of Solutions – Computational Approach, Ellis Horwood, London, 1995 Search PubMed.
- (a) E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS PubMed; (b) M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS; (c) V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404–417 CrossRef CAS.
- (a) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS PubMed; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- (a) A. Savin, A. D. Becke, J. Flad, R. Nesper, H. Preuss and H. G. Vonschnering, Angew. Chem., Int. Ed., 1991, 30, 409–412 CrossRef PubMed; (b) B. Silvi and A. Savin, Nature, 1994, 371, 683–686 CrossRef CAS PubMed; (c) A. Savin, B. Silvi and F. Colonna, Can. J. Chem., 1996, 74, 1088–1096 CrossRef CAS; (d) A. Savin, R. Nesper, S. Wengert and T. F. Fassler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1808–1832 CrossRef CAS PubMed.
- S. Noury, X. Krokidis, F. Fuster and B. Silvi, Comput. Chem., 1999, 23, 597–604 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Revision A.02, Gaussian Inc, Wallingford CT, 2009 Search PubMed.
- (a) R. G. Parr and W. Yang, Annu. Rev. Phys. Chem., 1995, 46, 701–728 CrossRef CAS PubMed; (b) H. Chermette, J. Comput. Chem., 1999, 20, 129–154 CrossRef CAS; (c) F. De Proft and P. Geerlings, Chem. Rev., 2001, 101, 1451–1464 CrossRef CAS PubMed; (d) P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1873 CrossRef CAS PubMed; (e) P. W. Ayers, J. S. M. Anderson and L. J. Bartolotti, Int. J. Quantum Chem., 2005, 101, 520–534 CrossRef CAS PubMed; (f) J. L. Gázquez, J. Mex. Chem. Soc., 2008, 52, 3–10 Search PubMed; (g) R. F. Nalewajski, J. Korchowiec and A. Michalak, Density Functional Theory IV, Topics in Current Chemistry, Springer, Berlin, Heidelberg, 1996, vol. 183, p. 25 Search PubMed; (h) P. Geerlings, S. Fias, Z. Boisdenghien and F. De Proft, Chem. Soc. Rev., 2014, 43, 4989–5008 RSC.
- R. G. Parr, L. von Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- (a) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS; (b) R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- (a) L. R. Domingo, E. Chamorro and P. Pérez, J. Org. Chem., 2008, 73, 4615–4624 CrossRef CAS PubMed; (b) L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2011, 9, 7168–7175 RSC.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, 1133–1138 CrossRef.
- L. R. Domingo, P. Pérez and J. A. Sáez, RSC Adv., 2013, 3, 1486–1494 RSC.
- D. H. Ess, G. O. Jones and K. N. Houk, Adv. Synth. Catal., 2006, 348, 2337–2361 CrossRef CAS PubMed.
- L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, Tetrahedron, 2002, 58, 4417–4423 CrossRef CAS.
- L. R. Domingo, P. Pérez and J. A. Sáez, Tetrahedron, 2013, 69, 107–114 CrossRef CAS PubMed.
- W. Benchouk, S. M. Mekelleche, B. Silvi, M. J. Aurell and L. R. Domingo, J. Phys. Org. Chem., 2011, 24, 611–618 CrossRef CAS PubMed.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- A. Savin, J. Chem. Sci., 2005, 117, 473–475 CrossRef CAS.
- B. Silvi, J. Mol. Struct., 2002, 614, 3–10 CrossRef CAS.
- (a) L. R. Domingo and J. A. Sáez, RSC Adv., 2014, 4, 58559–58566 RSC; (b) L. R. Domingo, M. Ríos-Gutiérrez and J. A. Sáez, RSC Adv., 2015, 5, 37119–37129 RSC.
- R. B. Woodward and R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1969, 8, 781–853 CrossRef CAS PubMed.
- K. N. Houk, J. Gonzalez and Y. Li, Acc. Chem. Res., 1995, 28, 81–90 CrossRef CAS.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry. Part A: Structure and Mechanisms, Springer, New York, 2000 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: MPW1K/6-31G(d) total electronic energies, in gas phase and in DCM, and B3LYP/6-31G(d) gas phase total and relative electronic energies of the stationary points involved in the stereo- and regioisomeric reaction paths of the 32CA reaction between nitrone 15 and acrolein 16. B3LYP/6-31G(d) geometries of the TSs involved in the studied 32CA reaction. MPWB1K/6-31G(d,p) cartesian coordinates, in gas phase, of the whole stationary points found. See DOI: 10.1039/c5ra10458k |
| This journal is © The Royal Society of Chemistry 2015 |