DOI:
10.1039/C5RA10455F
(Paper)
RSC Adv., 2015,
5, 62697-62705
Preparation of molecularly imprinted polymers using ion-pair dummy template imprinting and polymerizable ionic liquids†
Received
3rd June 2015
, Accepted 3rd July 2015
First published on 6th July 2015
Abstract
Ionic liquid based molecularly imprinted polymers have attracted considerable attention as biomimetic recognition materials due to their water-compatibility and high binding capacities. However, the selective recognition was unsatisfactory. In order to overcome this defect, we developed a novel dummy template ionic liquid based molecularly imprinted polymer, which used 1-butyl-3-vinylimidazolium α-aminohydrocinnamic acid salt as a functional monomer and the dummy template. Binding experiments showed that the obtained molecularly imprinted polymer possesses a high binding capacity (280.18 μmol g−1), imprinting factor (3.17) and selectivity factor (5.75). Molecular simulation results demonstrated that the high selectivity is attributed to the formation of ion-pairs between imidazolium and L-phenylalanine, which could be located in the imprinted cavities to improve the imprinted material’s efficiency. Subsequently, the dummy template ionic liquid based imprinted polymer was employed as packing in a solid phase extraction cartridge to analyze the L-phenylalanine in the blood of a phenylketonuria patient. The results indicated that the obtained dummy template ionic liquid based imprinted polymer has good analytical performance. Thus, the dummy imprinting combined with the ionic liquid is a useful way to improve the specific recognition of ionic liquid based molecular imprinted polymers, so that this method offers promising new applications in the field of the analysis of biological samples.
Introduction
Phenylketonuria (PKU) is a kind of genetic disease that causes a deficiency in phenylalanine hydroxylase.1 In people with PKU, L-Phe cannot be converted to tyrosine via the normal biochemical pathways and the L-Phe accumulates in plasma, leading to developmental delay and neurological damage.2 Thus, PKU screening and diagnosis is necessary for newborns. Conventional strategies include using polymerase chain reaction-single strand conformation polymorphism,3 denaturing gradient gel electrophoresis,4 bacterial growth inhibition assays,5 fluorometric immunoassays,6 tandem mass spectrometry,7 DNA hybridization,8 or enzymatic colorimetric assay methods.9 However, these methods can hardly meet the demand of clinical PKU screening in terms of analysis time, cost, reliability, etc. Therefore, rapid, low cost, efficient, and high accuracy methods for PKU screening are particularly important.
Molecularly imprinted technology (MIT) has been recognized as a powerful method and is also an interesting concept to generate polymer-based molecular recognition elements tailor-made for a given target or group of target molecules.10,11 Molecularly imprinted polymers (MIPs) are synthetic materials with artificially generated recognition sites able to rebind a specific target molecule, in preference to other closely-related compounds,12 and they have been applied in sensors and biosensors,13,14 solid-phase extraction,15 affinity chromatography,16–18 enzyme-like catalysts,19 enantioseparation,20,21 and drug delivery.22 In past decades, non-covalent MIT achieved great success in aprotic and low polarity organic solvents. However, the imprinting of hydrophilic biomolecules such as water-soluble amino acids remains a challenge due to their solubility, i.e. their insolubility in non-polar organic solvents or weakly polar solvents. Thus, very few functional monomers are available and polar solvents are not conducive to the stability of the template–monomer complex.23 To solve this problem, a strategy has been proposed: the conversion of the hydrophilic template to a hydrophobic one via alkyl chain attachment in a nonpolar or weakly polar solvent.24 It is worth noting that this uses an ion-pair as a dummy template. Alizadeh coupled sodium dodecyl sulfate to pyridoxine (a target molecule) using ion-pair formation, and then the ion-pair complex was used as a template to prepare MIPs for determining the water-soluble vitamin B6.25 Tominaga et al. used the ion-pair complex formed between a p-styrenesulfonic acid sodium salt and 4-(tributyl-ammonium-methyl)benzyltributylammonium as a template to prepare MIPs for the determination of the shellfish paralytic poison saxitoxin.26 These obtained MIPs showed improved selectivity. However, searching for new monomers with stronger interactions toward the template is the easiest and most direct way for imprinting water-soluble molecules.27
Recently, polymerizable ionic liquids (PILs) as an alternative functional monomer were considered by some researchers to prepare water-compatible MIPs.28–30 Usually, ionic liquid-based MIPs (IL-based MIPs) involve multiple interactions, such as hydrogen bonding, hydrophobic, π-stacking, electrostatic, and anion-exchange interactions. Therefore, the IL-based MIPs as novel materials have attracted the attention of researchers. Du et al. synthesized 1-(α-allylacetate)-3-vinylimidazolium chloride, and the obtained IL was employed as a functional monomer to prepare thymopentin imprinted microspheres via precipitation polymerization and surface imprinting techniques. The prepared MIPs had the capability of specific recognition for thymopentin from the thymopentin injection. There has even been a report of a L-Phe IL-based MIP.31 Guan et al. utilized 1-vinyl-3-carboxymethylimidazolium chloride ([VCIM]Cl) as a functional monomer to prepare MIPs for the selective recognition of L-Phe in a mixture of amino acids in solution. The obtained IL-based MIPs showed a definite selectivity for the template.32 However, while an anion-exchange interaction can significantly increase the capacity of IL-based MIPs, it can also reduce the selectivity for the target structures. Perhaps a counter ion, such as a bromide or chloride ion, as a hydrogen bond receptor would destroy the interactions between the template and the functional monomers. Thus, there would be a random distribution of the charged monomers on the surface of the IL-based MIPs instead of them being only located inside of the imprinted cavities.33 Probably, decreasing the disturbance of the counter-ion to improve the binding efficiency of the charged monomer and template should be a key factor.
In the present study, ion-pair dummy template imprinting and PIL were combined to prepare a L-Phe imprinted polymer on microspheres which contained cumyl dithiobenzate (a kind of reversible addition–fragmentation chain transfer agent, i.e. RAFT agent), which was needed in order to graft the MIP and NIP film from the base microspheres to be convenient for mass transfer. In detail, we designed a kind of novel amino acid IL: 1-butyl-3-vinylimidazolium α-aminohydrocinnamic acid salt ([Bvim][Phe]). The cation of the IL is a polymerizable imidazolium and the anion part is the dummy template L-Phe fragment. This acted as the dummy template and a functional monomer for preparing the L-Phe imprinted polymer (MIP1). The aim is to increase the selective recognition through enhancing the template–monomer interactions and avoiding disturbance of the anion. For comparison, the traditional IL-based L-Phe MIP was obtained using 1-butyl-3-vinylimidazolium bromide salt as the functional monomer (MIP2). Subsequently, the adsorption capacity of the MIPs was compared through adsorption kinetics, adsorption isotherms, competitive binding and molecular simulation results. Finally, the obtained MIPs were employed as packing material for solid phase extraction (SPE) to analyze L-Phe in the serum of PKU patients.
Experimental
Materials
Acetonitrile, methanol, and ethanol were provided by Tianjin Kermel Chemical Reagent Co., Ltd. L-Phe, L-His, L-Trp, and hydrochloric acid were obtained from the Sinopharm Chemical Reagent Co., Ltd. Divinylbenzene (DVB) and azobisisobutyronitrile (AIBN) were purchased from Tianjin Guangfu Fine Chemical Reagent Institute Co., Ltd. Ethylene dimethacrylate (EGDMA) and 4-vinylpyridine (4-VP) were purchased from J&K Scientific, Ltd. All chemicals were of analytical grade and were used without further purification. 1-Butyl-3-vinylimidazolium bromide ([Bvim]Br) and the 1-butyl-3-vinylimidazolium α-aminohydrocinnamic acid salt ([Bvim][Phe]) were obtained according to their synthetic methods in the literature,34,35 the 1H-NMR spectra are shown in Fig. S1 and S2 (ESI†). Cumyl dithiobenzate (CDB) was prepared following the literature method,36 the 1H-NMR spectrum is shown in Fig. S3 (ESI†). Human serum was collected from healthy volunteers and PKU patients, respectively. All experiments were performed in compliance with the relevant Chinese and institutional laws and guidelines.
Molecular simulations
All simulations were performed using Materials Studio 6.1 (Accelrys, San Diego, CA, USA). The template, functional monomers and solvent molecules were built using the Sketch tool. Subsequently, all of the components were randomly seeded into a cubic simulation box with periodic boundary conditions in all three orthogonal directions at a target density of 1.00 cm g−1. The components of the mixture are listed in Table 1. The periodic box was initially subjected to energy minimization using the Smart minimize method to eliminate the local non-equilibrium. The convergence level was set to 0.001 kcal mol−1 Å−1. Then molecular dynamic (MD) simulations were performed at 298 K for 1 ns to relax the system, in the NVT ensemble. In order to further relax the local hot-spots and to allow the system to achieve equilibrium, these structures were subjected to a 5-circle thermal annealing from 300 to 1000 K and then back to 300 K with 50 K intervals. At each temperature, a 100 ps NPT MD simulation was performed at a constant pressure (0.0001 GPa) with a time step of 1 fs. After the 5-circle annealing, a 100 ps NVT MD simulation was carried out at constant volume and a 250 ps NPT MD simulation at 0.0001 GPa and 298 K. Trajectories were saved every 1 ps and the final 50 ps configurations were used for analysis. During the whole simulation process, the temperature and pressure were maintained using the Berendsen method. The interatomic interactions were computed using the Condensed-Phase Optimized Molecular Potentials for Atomistic Simulation Studies (COMPASS) force field.37 The radial distribution functions (RDF, g(r)) were analyzed.
Table 1 Polymers studied in this experiment
| Polymers |
L-Phe |
[Bvim][Phe] amount (mmol) |
[Bvim]Br amount (mmol) |
4-VP amount (mmol) |
EGDMA amount (mmol) |
| MIP1 |
— |
0.20 |
— |
0.60 |
2.40 |
| MIP2 |
0.20 |
— |
0.20 |
0.60 |
2.40 |
| NIPs |
— |
— |
0.20 |
0.60 |
2.40 |
The interaction energy (ΔE) was evaluated using a script which defined the template molecules, functional monomers and template–monomers as sets in the trajectory files, respectively. Then, the potential energy of each set was calculated for each frame, and the ΔE was obtained through the equation:38
| | |
ΔE = E(template–monomer) − E(template) − E(monomer)
| (1) |
where
E(template–monomer) is the potential energy of the template–monomer complex.
E(template) and
E(monomer) are the potential energy of the template and the monomer in solvent, respectively.
Preparation of the MIPs and NIPs
0.05 wt% AIBN and CDB (1/3.5, w/w) were added into a one-neck flask containing a 2% (v/v) DVB acetonitrile solution (80.0 mL). The flask was immersed in a water bath under 90 °C for 6 h. The PDVB microspheres (those with the chain transfer agent contained on the surface of the PDVB were referred to as surface-CTA) were separated via vacuum filtration through a G-5 sintered glass filter and washed with ethanol three times. The microspheres were then dried at 50 °C in a vacuum oven until their weight was constant.
The template molecules and 4-VP were mixed in a one-neck flask and dissolved in 60 mL of acetonitrile–water (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), and incubated overnight in an oscillator at 25 °C. EGDMA, surface-CTA and AIBN were added to the above solution, purged with nitrogen for 30 min, and then sealed (with the compositions as indicated in Table 1). The flask was immersed in a constant-temperature water bath at 75 °C and stirred for 24 h. The generated polymer microspheres were collected by filtration, after which they underwent 5 h of Soxhlet extraction with a 10% acetic acid solution, methanol and drying at 50 °C in a vacuum. The corresponding non-imprinted microspheres (NIPs) were prepared and purified under identical conditions, but without the template addition.
:1 v/v), and incubated overnight in an oscillator at 25 °C. EGDMA, surface-CTA and AIBN were added to the above solution, purged with nitrogen for 30 min, and then sealed (with the compositions as indicated in Table 1). The flask was immersed in a constant-temperature water bath at 75 °C and stirred for 24 h. The generated polymer microspheres were collected by filtration, after which they underwent 5 h of Soxhlet extraction with a 10% acetic acid solution, methanol and drying at 50 °C in a vacuum. The corresponding non-imprinted microspheres (NIPs) were prepared and purified under identical conditions, but without the template addition.
Characterization of the chemical structure and morphology of MIPs/NIPs
A Fourier-transform infrared (FT-IR) spectrometer (Shimadzu, WQF-310, Japan) that uses the KBr compressed pellet method was used to identify the structures of the surface-CTA, MIPs and NIPs. The surface morphology of the MIPs, NIPs and surface-CTA were determined using scanning electron microscopy (FEI, Quanta 600FEG, USA). The surface property analysis was performed using nitrogen sorption porosimetry on a Surface Area and Porosimetry Analyzer (TriStarII 3020). The particle size distribution was measured using a laser diffraction technique (Mastersizer 2000, Malvern Instruments) in aqueous suspensions.
Binding, selectivity and real samples test
500 mg of MIPs or NIPs were immersed in 3 mL of a L-Phe standard aqueous solution at 25 °C. After filtration through an acetate membrane (pore size: 0.22 μm), the residual concentration of L-Phe was determined using a UV-Vis spectrophotometer (Shimadzu, UV-1750, Japan) at 257 nm, and the adsorption capacity (Q) was calculated using the following formula:where C0 is the initial L-Phe concentration, Ce is the residual concentration, V is the adsorption volume, and m is the amount of adsorbent.
Moreover, the imprinting factor (IF) was considered to calculate the recognition capabilities of the MIPs.
where
QMIPs is the amount of
L-Phe extracted by the MIPs, and
QNIPs is the amount of
L-Phe extracted by the NIPs in a similar process.
In order to estimate the recognition of the MIPs for L-Phe, L-His, L-Trp and D-Phe were chosen as competing molecules. In a typical procedure, 500 mg of MIPs or NIPs was incubated in 3.0 mL of an aqueous solution whose concentration of both L-Phe and the competing molecule was 378 μmol L−1 at 25 °C for 24 h. The concentration of L-Phe (L-His and L-Trp) was determined by HPLC. The column was a Kromasil CelluCoat (5 μm, 250 × 4.6 i.d.). The mobile phase was 10 mmol L−1 acetic acid aqueous solution–acetonitrile (30:70, v/v) and its flow rate was set at 0.7 mL min−1. The detection wavelength of the detector was set at 218 nm. The selective recognition factor (α) of the MIPs was calculated using the following equation:
where
QL-Phe and
QAnalogue are the amount of
L-Phe and competitor molecules extracted by the MIPs or NIPs in a similar process.
Assay of L-Phe in human serum
Empty stationary phase extraction (SPE) cartridges of 4 mL capacity were capped with fritted polypropylene disks at the bottom and on the top were packed with 700 mg of each polymer (MIPs and NIPs). Before each use, the sorbents were conditioned with acetonitrile (5 mL) followed by water (5 mL). For the MIP-SPE experiments, 5 mL of the human serum samples (the plasma was diluted five times with acetonitrile and centrifuged at 3000 rpm for 10 min) free from analytes were filtered using 0.22 μm pore size cellulose filters and spiked with different amounts of L-Phe to reach a final concentration of 10, 40, 100, 200, 400, 600 and 1000 nmol mL−1. The samples were percolated through the MIP-SPE cartridge at a flow rate of 0.2 mL min−1. The sorbent was washed with 1 mL of methanol. Full vacuum was applied for 5 min to ensure that the polymer was completely dry. Then, the sorbent was washed with 0.5 mol mL−1 of hydrochloric acid (1 mL). The eluent solution was analysed using HPLC. The column was a Kromasil ODS C18 (5 μm, 250 × 4.6 i.d.). The mobile phase was an acetonitrile–phosphate buffer solution (20 mmol L−1, pH = 7.4, 3:97, v/v) and its flow rate was set at 0.7 mL min−1. The detection wavelength of the detector was set at 254 nm.
Results and discussion
Morphological characterization
The surface-CTA obtained via RAFT distillation precipitation polymerization shows a smooth surface and a fine spherical degree, with a microsphere diameter that is approximately equal to 4 μm (Fig. 1a). Compared with the SEM images of the surface-CTA, the surfaces of MIP1 (b), MIP2 (c) and the NIPs (d) are rough, proving that the NIPs/MIPs have bonded with new polymer films. The particle sizes of the MIPs and NIPs were 19.9 (MIP1), 20.1 (MIP2) and 20.5 (NIPs) μm (see Fig. S4 in the ESI†), respectively. To verify the chemical structure, FT-IR was used and the results are shown in Fig. S5 (see the ESI†). Fig. S5b–d† show the infrared spectra of MIP1, MIP2 and the NIPs, respectively. The peaks at 1738 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration), 1257, and 1156 cm−1 (C–O–C stretching vibration) supported the existence of poly(EGDMA) in the obtained MIPs and NIPs. The characteristic peaks corresponding to CC and CN stretching (1632, 1601, 1513–1451, and 1388 cm−1) in the phenyl, pyridine, and imidazole rings are also observed in the spectra, it indicated that MIP and NIP coatings were successfully prepared. In addition, the prepared MIPs and NIPs interestingly presented irregular agglomeration. The morphologies are similar to the core–shell Asn-MIP structure which was described in our previously reported work.39 This might be caused by the physical and chemical properties of ILs. An IL is a kind of organic salt which consists of only ions.40 A small amount of an IL increases the polarity of the pre-polymerization solution. Because the surface-CTA was prepared using DVB, which lead to a relatively hydrophobic surface, the hydrophobic interaction becomes stronger with increasing polarity. Thus, the IL-based MIPs and NIPs can undergo serious agglomeration. Moreover, the surface properties of the MIPs and NIPs were studied using BET analysis, and the investigation using BET found surface areas of 2.2 m2 g−1 (MIP1), 2.6 m2 g−1 (MIP2) and 2.4 m2 g−1 (NIPs), and average pore sizes of 128.3 Å (MIP1), 125.6 Å (MIP2) and 127.2 Å (NIPs). These results indicate that the morphology of the MIPs and NIPs are almost the same.
O stretching vibration), 1257, and 1156 cm−1 (C–O–C stretching vibration) supported the existence of poly(EGDMA) in the obtained MIPs and NIPs. The characteristic peaks corresponding to CC and CN stretching (1632, 1601, 1513–1451, and 1388 cm−1) in the phenyl, pyridine, and imidazole rings are also observed in the spectra, it indicated that MIP and NIP coatings were successfully prepared. In addition, the prepared MIPs and NIPs interestingly presented irregular agglomeration. The morphologies are similar to the core–shell Asn-MIP structure which was described in our previously reported work.39 This might be caused by the physical and chemical properties of ILs. An IL is a kind of organic salt which consists of only ions.40 A small amount of an IL increases the polarity of the pre-polymerization solution. Because the surface-CTA was prepared using DVB, which lead to a relatively hydrophobic surface, the hydrophobic interaction becomes stronger with increasing polarity. Thus, the IL-based MIPs and NIPs can undergo serious agglomeration. Moreover, the surface properties of the MIPs and NIPs were studied using BET analysis, and the investigation using BET found surface areas of 2.2 m2 g−1 (MIP1), 2.6 m2 g−1 (MIP2) and 2.4 m2 g−1 (NIPs), and average pore sizes of 128.3 Å (MIP1), 125.6 Å (MIP2) and 127.2 Å (NIPs). These results indicate that the morphology of the MIPs and NIPs are almost the same.
 |
| | Fig. 1 The SEM images of the surface-CTA (a), MIP1 (b), MIP2 (c) and the NIPs (d). | |
Binding properties of the MIPs and NIPs
Adsorption kinetics were carried out using 10 mmol L−1 L-Phe and the results are shown in Fig. 2. It can be seen that the adsorption capacity increased with time and that MIP1, MIP2 and the NIPs had a fast adsorption rate. In the first 45 min, the adsorption increased rapidly and achieved 90% of the equilibrium adsorption capacity. Subsequently, the adsorption rate was slower from 45 min to 135 min for MIP1 and 45–90 min for MIP2. The adsorption had almost reached an equilibrium after 135 min (MIP1) or 90 min (MIP2). Based on the above phenomenon, the adsorption kinetic curves of MIP1 and MIP2 can be obviously divided into three phases. The maximum slope was observed in the first stage, it indicated that the L-Phe in the aqueous solution was adsorbed onto the surface of MIP1 and MIP2 rapidly by electrostatic or other weak interactions. In the second period, the adsorption ratio was slower due to the fact that the adsorbed L-Phe diffused into the inner imprinted cavities of the surface of the MIPs. The diffusion was difficult because of the close match between L-Phe and the imprinting sites. However, this case does not exist on the NIPs due to the absence of imprinting sites matched with L-Phe on the surface of the NIPs and the fact that the distribution of functional groups on the surface of the NIPs was random.33 Since the adsorption equilibrium is achieved at the third stage, the slope approached zero. Moreover, the length of the diffusion time also greatly differs in the second stage. Compared to MIP2, the diffusion time of MIP1 is longer, the reason might be that MIP1 has a higher imprinting efficiency. At this point, it is consistent with simulation results.
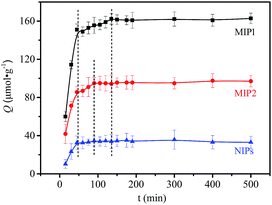 |
| | Fig. 2 Curve of adsorption kinetics for MIP1 (black line), MIP2 (red line) and the NIPs (blue line). | |
The binding isotherms of L-Phe to the MIPs and NIPs were determined in the concentration range of 1–100 mmol L−1 (initial concentration) and the results are shown in Fig. 3. It can be seen that the amount of L-Phe bound to the MIPs increased quickly with increasing initial concentration below 70 mmol L−1. However, when the initial concentration was above 70 mmol L−1, the adsorption curve became relatively flat and reached saturation. Moreover, from the amount of L-Phe bound to MIP1 (280.18 μmol g−1) and MIP2 (205.30 μmol g−1), the IFs were calculated to be 3.17 and 2.27, and were dramatically higher than those of the other L-Phe MIPs reported.41,42 This showed that the IL-based MIPs possessed high binding capacities and more accurate imprinting cavities.
 |
| | Fig. 3 Curve of the adsorption isotherms for MIP1 (black line), MIP2 (red line) and NIPs (blue line). | |
To obtain insight into the adsorption mechanism, equilibrium isotherm equations like the Langmuir isotherm (the one binding site model) and Freundlich isotherm (the continuous distribution model) are utilized to describe the experimental adsorption data, which can be expressed, respectively, as follows:
| | |
Ce/Q = 1/QmaxK + Ce/Qmax
| (5) |
where
Q (mmol g
−1) is the amount of
L-Phe bound to the MIPs and NIPs at equilibrium,
Qmax (mmol g
−1) is the apparent maximum adsorption capacity,
Ce (mmol L
−1) is the free analytical concentration at equilibrium,
K (L mmol
−1) is the affinity constant and
m are the Freundlich constants which represent the heterogeneity of the system, respectively. The values of
K,
Qmax and
m can be calculated from the slope and intercept of the linear plot of
Ce/
Q versus Ce and log
Q versus log
Ce, respectively.
The fitting results are shown in Fig. S6 (see the ESI†) and Table 2. The correlation coefficients (R2) for the Langmuir equation for all polymers were >0.997, and for the Freundlich were <0.94 respectively. Therefore, the Langmuir isotherm model is more suitable for the adsorption process due to the higher correlation coefficients (R2). It can be concluded that the adsorption of L-Phe onto the MIPs and NIPs may be a monolayer adsorption.43 Comparing the two kinds of IL-based MIPs, they are significantly different in their apparent maximum adsorption capacities and binding constants. For MIP1, both the Qmax and K are much better than MIP2 toward L-Phe, which might be caused by the anions of the ILs. For MIP2, the Br− probably disturbed the interaction between L-Phe and the functional monomer during the pre-polymerization stage, and thus the prepared IL-based MIPs possess a lower affinity toward the template. For the ionized template, the anion of the IL can avoid this interference and improve the imprinting efficiency for MIP1. This view is also supported by the molecular simulations.
Table 2 Equations and parameters of the adsorption isotherms of the MIPs and NIPs
| Model |
Equation and parameters |
MIP1 |
MIP2 |
NIPs |
| Langmuir isotherm |
Equation |
Ce/Q = 5.668811 + 3.47560Ce |
Ce/Q = 21.907710 + 4.52429Ce |
Ce/Q = 121.329950 + 9.21535Ce |
| Qmax (μmol g−1) |
287.72 |
221.03 |
108.51 |
| K (L mmol−1) |
0.61 |
0.21 |
0.076 |
| R2 |
0.9989 |
0.9989 |
0.9979 |
| Freundlich isotherm |
Equation |
logQ = 0.1679logCe − 0.824 |
logQ = 0.250logCe − 1.114 |
logQ = 0.3788logCe − 1.715 |
| K (L mmol−1) |
0.15 |
0.077 |
0.019 |
| m |
0.1679 |
0.2497 |
0.3788 |
| R2 |
0.8864 |
0.9496 |
0.8895 |
Selectivity and recognition
The binding selectivity of MIP1 in aqueous solution was evaluated by measuring the competitive binding capacities towards L-Phe, L-His, L-Trp and D-Phe (for the molecular structures see Scheme 1) since they had similar chemical structures and isoelectric points (L-Phe pI = 5.48, L-His pI = 7.59, L-Trp pI = 5.89 and D-Phe pI = 5.48). For comparison, the MIP2 and NIP were chosen as reference polymers. Fig. 4 shows the results of the competitive bindings of L-Phe and competitors on these three polymers. Surprisingly, the specific template binding abilities (i.e., the binding differences between the template and the competitors) of the NIPs almost completely disappear (α is in the range of 0.62 to 1.17) and the MIP2 shows poor selectivity for L-Phe (α is in the range of 1.18 to 1.57). In sharp contrast, the MIP1 shows unique specific template bindings and high selectivity for L-Phe (αD-Phe is 1.42, αL-His is 2.34 and αL-Trp is 2.20), which is probably attributed to the mode of monomer–template interactions during the imprinting process.
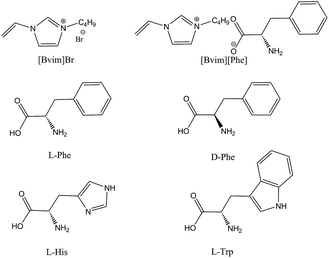 |
| | Scheme 1 Molecular structures of [Bvim]Br, [Bvim][Phe], L-Phe, D-Phe, L-His and L-Trp. | |
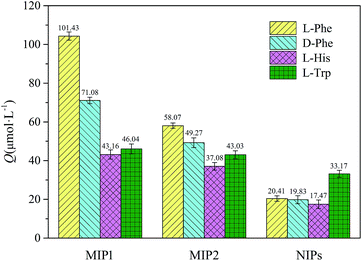 |
| | Fig. 4 Results of the competitive binding tests for the MIPs and NIPs with L-Phe (yellow column), D-Phe (light cyan column), L-His (light magenta column) and L-Trp (olive column). | |
In order to understand the process of imprinting, a comprehensive theoretical analysis of the various interactions existing in the pre-polymerization solution was undertaken. During the imprinting process, hydrogen bonding interactions play a significant role in the naturally occurring formation of a complex with high stability.44 In general, the hydrogen bonding was involved in Coulombic interactions and van der Waals forces. It is assumed that hydrogen bonding occurs as the donor–acceptor distance is smaller than 3.50 Å. Thus, the radial distribution functions (RDFs) were used to analyze the distances of the close contacts in the system. It describes how density varies as a function of distance from a reference particle.45 In Fig. 5A, the first peak was observed at 2.20 Å (MIP1). However, the sharp peak of 4-VP and L-Phe did not appear in the radial distribution function diagram for MIP2. These results indicate that the N atom of the 4-VP molecule preferentially interacts with the –NH2 of the L-Phe in MIP1, and the close contact of 4-VP and the L-Phe in MIP1 is more close than in MIP2. Fig. 5B presents the RDFs of the imidazolium and the template in MIP1 and MIP2. The first sharp peak appeared at 4.20 (MIP1) and 4.66 Å (MIP2), respectively. In MIP1, the peak of the imidazolium and the template was stronger and closer than in MIP2. The RDFs of imidazolium with the bromide anions was counted in MIP2 (see Fig. 5C), and the peaks were located at 4.53 Å, which indicated that there are ion-pairs formed between imidazolium and bromide. Moreover, the peak of L-Phe and the bromide ion was found in the RDF diagram for MIP2, showing that the interaction of the monomer and template has been disturbed by the bromide ion. All of these results demonstrate that the traditional IL-based MIPs with low selective recognition for the template might be ascribed to inefficient conjugation of the template and IL.
 |
| | Fig. 5 Radial distribution functions calculated for L-Phe and 4-VP (A), L-Phe and the imidazolium cations (B), and L-Phe/imidazolium and the bromide ions (C) in MIP2. | |
To further understand the interaction strength between the template and the functional monomers, we have quantitatively investigated the ΔE. In theory, a functional monomer giving a high binding energy with the template molecule would interact strongly with template molecules.46 In other words, when ΔE is higher, more easy adsorption is expected of the MIPs, which helps MIPs to recognize the template. In order to investigate the selectivity of MIP1 and MIP2, the binding energies of the template–monomer complexes were evaluated. In order to facilitate comparison, the mean values of ΔE between L-Phe and the functional monomers were calculated and are listed in Table 3. From Table 3, it can be seen that the ΔEImidazolium–VP are −15.43 (MIP1) and −10.07 (MIP2) kcal mol−1, respectively. The strongest interaction of the imidazolium–VP complex occurred in the MIP1 model. Similarly, the highest interaction energy for Phe–imidazolium was found in MIP1. Moreover, the ΔE of the Phe–Br− is −5.54 kcal mol−1 in MIP2. Compared to MIP1, all of the ΔE values of the template–monomer complexes are lower for MIP2. These results strongly suggest the random distribution of the charged monomers on the surface of the traditional IL-based MIPs instead of being located in the inside of the imprinted cavities. Therefore, an IL as the dummy template would be a promising method to avoid interference and to further increase the specific recognition of IL-based MIPs.
Table 3 Average ΔE of the interactions of L-Phe with the functional monomers in the pre-polymerization mixture
| MIP1 |
MIP2 |
| Functional monomers |
ΔE (kcal mol−1) |
Functional monomers or interference |
ΔE (kcal mol−1) |
| 4-VP |
−23.92 |
4-VP |
−19.14 |
| Imidazolium cations |
−15.43 |
Imidazolium cations |
−10.07 |
| — |
— |
Br− |
−5.54 |
Assay of L-Phe in human serum
The aim of this study is to investigate the feasibility of the analytical performance of the prepared MIP1 for the determination of L-Phe in human blood. The prepared MIPs and NIPs were packed into a solid-phase extraction cartridge to evaluate their characteristics. The chromatograms of the serum from a PKU patient with a L-Phe concentration of 400 nmol mL−1 adsorbed by the MIP and the NIP columns are displayed in Fig. 6. Comparing the chromatogram of MIP1-SPE with MIP2-SPE, MIP1 displayed a higher pre-concentration ability. In addition, all of the polymers showed good analytical performance, with correlation coefficients, R2, between 0.9931 and 0.9987 (Table 4). The precision was determined over five separations of L-Phe from the extract, the RSD was 3.5–4.1%. Based on a signal-to-noise ratio of 3, the limit of determination (LOD) for L-Phe was 3.0 nmol mL−1 on MIP1, which was 7 times lower than that on MIP2. This can be attributed to the fact that MIP1 had a much higher imprinting efficiency in aqueous media. All of these results demonstrate that there is a potential for dummy template IL-based MIPs to be widely applied to the determination of biomolecules.
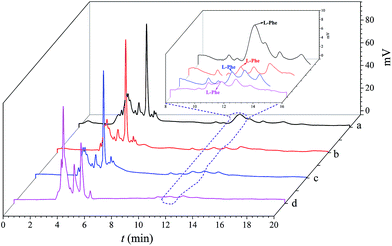 |
| | Fig. 6 Chromatograms of the human serum of PKU patients (d, magenta line), enriched with MIP1 (a, black line), MIP2 (b, red line) and NIPs (c, blue line). | |
Table 4 Linear range, LODs and repeatability of the method
| Polymer |
Linear range (nmol mL−1) |
R2 |
RSD (%) |
LOD (nmol mL−1) |
| MIP1 |
10.0–1000 |
0.9987 |
2.6 |
3.0 |
| MIP2 |
40.0–1000 |
0.9952 |
3.1 |
23.0 |
| NIPs |
40.0–1000 |
0.9931 |
3.4 |
20.0 |
Conclusions
We have successfully designed a novel kind of PIL, which was employed as a dummy template and a functional monomer to prepare a L-Phe molecularly imprinted polymer. Firstly, the binding tests show that the obtained MIP1 had a high adsorption capacity and high selectivity for binding L-Phe. Subsequently, the MD simulation results demonstrated that the hydrogen bonding of the template–monomer complex would be seriously damaged by the anion of a traditional PIL when it acts as a functional monomer directly. Thus, many of the charged monomers will be randomly distributed over the polymer surface, as well as being specifically located in the imprinted sites. Finally, the prepared MIP1 was used to bind the L-Phe in human serum. The MIP1 possess stronger binding ability than traditional IL-based MIPs and NIPs, therefore, MIP1 has a lower LOD and a wider linear range for determination of L-Phe in real biological samples. All of these results indicated that the anion of the IL as the dummy template is a useful way to improve the specific recognition of MIPs, and the versatility of the obtained MIPs can potentially be extended to a broad scope for effective PKU screening efforts in clinical laboratories.
Acknowledgements
The authors are grateful for the financial support provided by the National Nature Science Foundation of China (Nos 21174111 and 51433008). This paper was supported by the Center for High Performance Computing of Northwestern Polytechnical University, China.
Notes and references
- J. J. Mitchell, Y. J. Trakadis and C. R. Scriver, Genet. Med., 2011, 13, 697–707 CrossRef CAS PubMed.
- A. Turki, K. Ueda, B. Cheng, A. Giezen, S. Stockler and R. Elango, FASEB J., 2015, 29, 742–749 Search PubMed.
- A. Acosta, W. Silva Jr, T. Carvalho, M. Gomes and M. Zago, Hum. Mutat., 2001, 17, 122–130 CrossRef CAS.
- H. G. Eiken, Hum. Mutat., 1996, 8, 19–22 CrossRef CAS.
- H. Karamifar, Iran. J. Pediatr., 2010, 20, 216–220 Search PubMed.
- S. Khemir, Psychiatr., Neurol., Neurochir., 2011, 113, 727–730 Search PubMed.
- D. M. Niu, Y. H. Chien, C. C. Chiang, H. C. Ho, W. L. Hwu, S. M. Kao, S. H. Chiang, C. H. Kao, T. T. Liu, H. Chiang and K. J. Hsiao, J. Inherited Metab. Dis., 2010, 33, 295–305 CrossRef PubMed.
- B. Chen, X. Zhou, C. Li, Q. Wang, D. Liu and B. Lin, J. Chromatogr. A, 2011, 1218, 1907–1912 CrossRef CAS PubMed.
- G. Thiessen, R. Robinson, K. De Los Reyes, R. J. Monnat and E. Fu, Analyst, 2015, 140, 609–615 RSC.
- H. Qiu, Y. Xi, F. Lu, L. Fan and C. Luo, Spectrochim. Acta, Part A, 2012, 86, 456–460 CAS.
- D. F. Tai, C. Y. Lin, T. Z. Wu and L. K. Chen, Anal. Chem., 2005, 77, 5140–5143 CrossRef CAS PubMed.
- A. M. Esteban, Trends Anal. Chem., 2013, 45, 169–181 CrossRef PubMed.
- T. Alizadeh, M. Zare, M. R. Ganjali, P. Norouzi and B. Tavana, Biosens. Bioelectron., 2010, 25, 1166–1172 CrossRef CAS PubMed.
- G. Mustafa and P. A. Lieberzeit, RSC Adv., 2014, 4, 12723–12728 RSC.
- S. Wei, Y. Liu, Z. Yan and L. Liu, RSC Adv., 2015, 5, 20951–20960 RSC.
- F. Meier, B. Schott, D. Riedel and B. Mizaikoff, Anal. Chim. Acta, 2012, 744, 68–74 CrossRef CAS PubMed.
- J. Haginaka, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 866, 3–13 CrossRef CAS PubMed.
- S. Hajizadeh, C. Xu, H. Kirsebom, L. Ye and B. Mattiasson, J. Chromatogr. A, 2013, 1274, 6–12 CrossRef CAS PubMed.
- W. Chen, D. K. Han, K. D. Ahn and J. M. Kim, Macromol. Res., 2002, 10, 122–126 CrossRef CAS.
- J. J. Torres, N. Gsponer, C. L. Ramírez, D. M. A. Vera, H. A. Montejano and C. A. Chesta, J. Chromatogr. A, 2012, 1266, 24–33 CrossRef CAS PubMed.
- P. Qu, J. Lei, L. Zhang, R. Ouyang and H. Ju, J. Chromatogr. A, 2010, 1217, 6115–6121 CrossRef CAS PubMed.
- P. Wang, A. Zhang, Y. Jin, Q. Zhang, L. Zhang, Y. Peng and S. Du, RSC Adv., 2014, 4, 26063–26073 RSC.
- S. Xu, H. Lu and L. Chen, J. Chromatogr. A, 2014, 1350, 23–29 CrossRef CAS PubMed.
- P. Manesiotis, A. J. Hall, J. Courtois, K. Irgum and B. Sellergren, Angew. Chem., Int. Ed., 2005, 117, 3970–3974 CrossRef PubMed.
- T. Alizadeh, Anal. Chim. Acta, 2008, 623, 101–108 CrossRef CAS PubMed.
- Y. Tominaga, T. Kubo, K. Kaya and K. Hosoya, Macromolecules, 2009, 42, 2911–2915 CrossRef CAS.
- L. Chen, S. Xu and J. Li, Chem. Soc. Rev., 2011, 40, 2922–2942 RSC.
- W. Bi, M. Tian and K. H. Row, J. Chromatogr. A, 2012, 1232, 37–42 CrossRef CAS PubMed.
- L. Qian, X. Hu, P. Guan, B. Gao, J. Li, C. Wang and Y. Tang, Talanta, 2014, 121, 56–64 CrossRef CAS PubMed.
- J. P. Fan, Z. Y. Tian, S. Tong, X. H. Zhang, Y. L. Xie, R. Xu, Y. Qin, L. Li, J. H. Zhu and X. K. Ouyang, Food Chem., 2014, 141, 3578–3585 CrossRef PubMed.
- C. Du, X. Hu, P. Guan, L. Guo, L. Qian, R. Song, J. Li and C. Wang, J. Mater. Chem. B, 2015, 3, 3044–3053 RSC.
- B. Gao, P. Guan, X. Hu, L. Qian, Q. Wang, L. Yang and D. Wang, Des. Monomers Polym., 2014, 18, 1–14 Search PubMed.
- M. J. Whitcombe, I. Chianella, L. Larcombe, S. A. Piletsky, J. Noble, R. Porter and A. Horgan, Chem. Soc. Rev., 2011, 40, 1547–1571 RSC.
- Z. H. Si, L. H. Qiu, H. L. Dong, F. L. Gu and F. Yan, ACS Appl. Mater. Interfaces, 2014, 6, 4346–4355 CAS.
- K. Fukumoto, M. Yoshizawa and H. Ohno, J. Am. Chem. Soc., 2005, 127, 2398–2399 CrossRef CAS PubMed.
- Y. Liu, J. He, J. Xu, D. Fan, W. Tang and Y. Yang, Macromolecules, 2005, 38, 10332–10335 CrossRef CAS.
- B. Qiao, X. Zhao, D. Yue, L. Zhang and S. Wu, J. Mater. Chem., 2012, 22, 12339–12348 RSC.
- L. Wu, K. Zhu, M. Zhao and Y. Li, Anal. Chim. Acta, 2005, 549, 39–44 CrossRef CAS PubMed.
- J. Li, X. Hu, P. Guan, R. Song, X. Zhang, Y. Tang, C. Wang and L. Qian, React. Funct. Polym., 2015, 88, 8–15 CrossRef CAS PubMed.
- R. Gao, X. Shangguan, G. Qiao and J. Zheng, Electroanalysis, 2008, 20, 2537–2542 CrossRef CAS PubMed.
- H. Wu, Y. Zhao, M. Nie and Z. Jiang, Sep. Purif. Technol., 2009, 68, 97–104 CrossRef CAS PubMed.
- Z. Zhang, M. Zhang, Y. Liu, X. Yang, L. Luo and S. Yao, Sep. Purif. Technol., 2012, 87, 142–148 CrossRef CAS PubMed.
- R. Gao, X. Mu, Y. Hao, L. Zhang, J. Zhang and Y. Tang, J. Mater. Chem. B, 2014, 2, 1733–1741 RSC.
- C. Dong, X. Li, Z. Guo and J. Qi, Anal. Chim. Acta, 2009, 647, 117–124 CrossRef CAS PubMed.
- J. P. Hansen and I. R. McDonald, Theory of Simple Liquids, Elsevier, 2nd edn, 1990 Search PubMed.
- M. S. Khan, P. S. Wate and R. J. Krupadam, J. Mol. Model., 2012, 18, 1969–1981 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra10455f |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 






