
Expanded graphite/phenolic resin-based carbon composite adsorbents for post-combustion CO2 capture†
Abstract
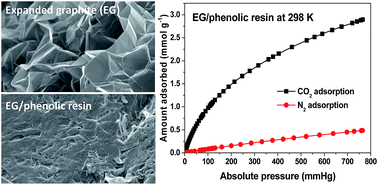
Porous carbon composite adsorbents were prepared from a commercial phenolic resin mixed with a small proportion of thermally expanded graphite (EG) followed by carbonization and physical activation with CO2. The addition of EG dramatically hastens the CO2 activation and results in remarkably enhanced microporosity development in the EG composite compared to the activated phenolic resin alone. The resultant EG composite adsorbents exhibit high CO2 adsorption capacities at 298 K and excellent CO2/N2 adsorption selectivity. In particular, the EG composite shows superior CO2 uptake at low CO2 pressures (47 mg g−1 at 298 K and 0.15 bar), which is more important to actual flue gas applications in post-combustion capture (PCC). Moreover, EG composite adsorbents are especially attractive as the EG component is inexpensive, available in very large amounts and easy to handle and is only required at a low addition level of around 2 wt%. The rapid CO2 activation and the low burn-off for excellent CO2 adsorption performance at low pressures greatly reduce the energy required to produce the adsorbent and the waste generated in activation. This further enhances the cost and environmental advantages of physically activated EG composites over those PCC adsorbents prepared by chemical activation and functionalization of porous carbons. Hence, due to its superior CO2 adsorption properties and favourable fabrication process, the newly developed EG carbon composite adsorbent holds great promise for large-scale deployment and commercial applications to PCC.
 Please wait while we load your content...
Please wait while we load your content...

