
Modification of a thin layer of α-Fe2O3 onto a largely voided TiO2 nanorod array as a photoanode to significantly improve the photoelectrochemical performance toward water oxidation†
Abstract
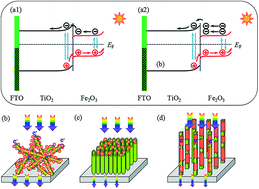
A largely voided TiO2 nanorod array was synthesized and further modified with a thin layer of α-Fe2O3 (Fe2O3@TiO2), by the pyrolysis of an FeCl3 ethanol solution, as a photoanode toward water oxidation, showing significantly improved photoelectrochemical performance over a TiO2 nanorod array. Among all of the Fe2O3 decorated TiO2-based photoanodes, the optimal voided Fe2O3@TiO2 nanorod array photoanode delivered the largest photocurrent density of 3.39 mA cm−2 at 1.23 V (vs. RHE) and the highest applied bias photon-to-current efficiency (ABPE) (1.153%) under 100 mW cm−2 UV-vis light illumination. In particular, the ABPE for the as-prepared photoanode was ∼3.3 times higher than that of the plain TiO2 nanorod array (0.35%), ∼11.3 times higher than that of the Fe2O3-modified randomly arranged TiO2 nanorods and ∼6.2 times higher than that of a Fe2O3-modified densely arranged TiO2 nanotube array. The significant enhancement mainly originates from the large voids in the nanorod array allowing a thin layer of Fe2O3 to fully modify the TiO2 nanorods, which improves the absorption of UV light, boosts the charge interface transfer rate, reduces the charge diffusion length and suppresses the charge recombination process. This work demonstrates a feasible route to improving the photoelectrochemical catalytic performance of TiO2 semiconductors toward water splitting.
 Please wait while we load your content...
Please wait while we load your content...

