
Fabrication and evaluation of protein imprinted polymer based on magnetic halloysite nanotubes
Xiaohong Zhua,
Hui Lia,
Hui Zhoub and
Shian Zhong*a
aSchool of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China. E-mail: zhongshian@aliyun.com; Tel: +86 13107210768
bThe Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha 410013, China
First published on 28th July 2015
Abstract
A novel protein imprinted polymer, which combined the surface imprinting technology and magnetic halloysite nanotubes (MHNTs), was prepared for selective separation of a template protein. In this work, MHNTs were synthesized through the coprecipitation method based on encapsulation of Fe3O4 nanoparticles in halloysite nanotubes. The surface of the MHNTs was modified with vinyl groups through reaction with 3-(methacryloyloxy)propyl trimethoxysilane. Subsequently, the modified MHNTs were used as a support, N-isopropylacrylamide and methacrylic acid as bifunctional monomers, N,N′-methylene bisacrylamide as the crosslinking agent, and ammonium persulphate and N,N,N′,N′-tetramethylethylenediamine as initiators. The resulting polymer was named MHNTs@BSA-MIP, which was characterized by transmission electron microscopy, Fourier transform infrared spectroscopy, X-ray diffraction, and vibrating sample magnetometry. The MHNTs@BSA-MIP exhibited high adsorption (48.4 mg g−1), good selectivity, rapid kinetic binding (45 min), fast magnetic separation (10 s), and favorable reproducibility (relative standard deviation <8% for batch-to-batch evaluation). The prepared MHNTs@BSA-MIP is suitable for separation and provides a significant reference for other proteins in proteomics.
1 Introduction
Recently, separation and enrichment of purified proteins have attracted considerable interest because of their increasing significance in various applications ranging from diagnostics to therapeutics. Traditionally, immobilized metal ion affinity chromatography,1 aqueous two-phase systems,2 or electrophoretic–electroosmotic focusing3 can be used to separate proteins from cell debris or to purify proteins from other proteins. However, these techniques have several limitations such as poor stability, high cost, non-reusability, and complex operation technology. Therefore, developing a new method for protein separation is of great importance.Molecular imprinting technology is a promising strategy for rapid and inexpensive production of biomimetic materials, which can generate synthetic materials with specific recognition sites.4,5 Compared with its biological counterparts, molecularly imprinted polymer (MIP) displays significant advantages, including high mechanical properties, chemical stability, easy preparation,6 potential reusability, and low manufacturing cost.7 Nevertheless, when regard to biological macromolecule imprinting, some inherent limitations exist, such as insolubility in water, poor mass transfer, and low integrity.8 In order to overcome these limitations, several strategies, including the use of metal-coordination polymerization,9,10 epitope approach,11–13 and surface imprinting,14–22 have been employed in protein imprinting.
Among these methods, surface imprinting with supporter has become an effective solution to achieve excellent recognition and adsorption, by which recognition sites were formed on the material surface. Different supporting substrates such as silica nanoparticles,23–25 carbon nanotubes (CNTs),26,27 Fe3O4,28,29 and oxidized metal complex nanoparticles, have been developed to prepare MIP. Great efforts have been performed to combine CNTs with the MIP. Chen et al. synthesized a novel MIP based on modified CNTs by using bovine serum albumin (BSA) as the template, which exhibited high sensitivity, and good reproducibility.30 Huang and his co-workers combined the advantages of CNT–AuNP composites and chitosan to design a disposable and sensitive MIP for tyramine detection.31
However, CNTs show poor dispersibility in organic phase or aqueous phase, and thus modification is necessary prior to use. By contrast, halloysite nanotubes (HNTs) disperse well in water. HNTs are aluminosilicate clay mineral with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Al:Si ratio and ideal chemical formula of Al2Si2O5(OH)4·nH2O, which were chemically similar to kaolin, but differ in having a hollow tubular structure rather than a stacked plate-like structure.32,33 HNTs exhibit potential application owing to their gibbsite octahedral sheet aluminum (innermost) and silicate (outermost) surfaces, which allow different chemical reactions in the interior and exterior surfaces. Similar to most natural materials, the sizes of HNTs vary between 500 and 1000 nm in length and 10–150 nm in inner diameter depending on the deposits. HNTs are environmentally friendly, and available at low cost. In addition, large reserves of HNTs are found in China.34 HNTs have unique properties including non-swelling, large surface area and abundant hydroxyl groups. Thus, the combination of Fe3O4 and HNTs can form the magnetic HNTs (MHNTs),35,36 which can be applied in adsorption technology. Pan and co-workers used MHNTs combined with MIP as sorbent for solid-phase extraction and separation of 2,4,6-trichlorophenol from environmental water samples.37 Our group also fabricated the MIP on the surface of MHNTs for recognition of herbicides.38 In these previous studies, low-molecular weight compounds were used as template molecules. However, no reports had described a preparation of protein-imprinted polymer on the surface of MHNTs. Therefore, we developed a new preparation method for covering MHNTs with MIP, combining both surface molecular imprinting technology and nanotechnology.
:1 Al:Si ratio and ideal chemical formula of Al2Si2O5(OH)4·nH2O, which were chemically similar to kaolin, but differ in having a hollow tubular structure rather than a stacked plate-like structure.32,33 HNTs exhibit potential application owing to their gibbsite octahedral sheet aluminum (innermost) and silicate (outermost) surfaces, which allow different chemical reactions in the interior and exterior surfaces. Similar to most natural materials, the sizes of HNTs vary between 500 and 1000 nm in length and 10–150 nm in inner diameter depending on the deposits. HNTs are environmentally friendly, and available at low cost. In addition, large reserves of HNTs are found in China.34 HNTs have unique properties including non-swelling, large surface area and abundant hydroxyl groups. Thus, the combination of Fe3O4 and HNTs can form the magnetic HNTs (MHNTs),35,36 which can be applied in adsorption technology. Pan and co-workers used MHNTs combined with MIP as sorbent for solid-phase extraction and separation of 2,4,6-trichlorophenol from environmental water samples.37 Our group also fabricated the MIP on the surface of MHNTs for recognition of herbicides.38 In these previous studies, low-molecular weight compounds were used as template molecules. However, no reports had described a preparation of protein-imprinted polymer on the surface of MHNTs. Therefore, we developed a new preparation method for covering MHNTs with MIP, combining both surface molecular imprinting technology and nanotechnology.
In this study, a facile and versatile approach was developed to prepare MIP based on modified MHNTs. First, MHNTs were fabricated by a straightforward and effective coprecipitation method. Then, a vinyl group was introduced on the surface of MHNTs to graft MIP onto MHNTs. Subsequently, MHNTs@BSA-MIP was synthesized by a precipitation polymerization method, in which the modified MHNTs were used as the support matrix, N-isopropylacrylamide (NIPAM) and methacrylic acid (MAA) as the co-monomers, N,N′-methylene bisacrylamide (BIS) as the crosslinking agent. Characterization and performance evaluation of the prepared MHNTs@BSA-MIP were conducted. The MHNTs@BSA-MIP can selectively recognize the template protein in a complex matrix and it possesses numerous imprinted cavities within the polymer network owing to the high surface-to-volume ratio of MHNTs.
2 Experimental
2.1 Chemicals and reagents
All chemicals were of analytical reagent grade. Double-distilled water was used in the experiments. The proteins were as follows: bovine serum albumin (BSA, MW = 68 kDa, pI = 4.9), lysozyme (Lyz, MW = 13.4 kDa, pI = 11.2), ovalbumin (OVA, MW = 43.0 kDa, pI = 4.7), and trypsin from bovine pancreas (Try, MW = 12.6 kDa, pI = 7.8). All proteins were purchased from Energy Chemical.HNTs were supplied by Zhengzhou Jinyangguang Co., Ltd (Henan, China). FeCl3·6H2O, FeCl2·4H2O, and 3-methacryloxypropyltrimethoxysilane (MPTES) were supplied by Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). NIPAM, MAA, BIS, ammonium persulphate (APS), and N,N,N′,N′-tetramethylethylenediamine (TEMED) were purchased from Aladdin Company (Shanghai, China). Sodium dodecyl sulfate (SDS) was purchased from Tianjin Chemical Reagents Co., Ltd.
2.2 Apparatus and conditions
UV-vis spectra were obtained using a UV-2450 Spectrophotometer (Shimadzu, Japan). Fourier transform infrared (FT-IR) spectroscopy was employed to obtain infrared spectra (Nicolet, America). The samples were prepared by mixing the products with KBr and pressing into a compact pellet. Morphology and size distribution of the MHNTs and MHNT@MIP were investigated using transmission electron microscopy (TEM, JEM-2100F, Japan) at 20.0 kV. Particle phases were characterized using X-ray diffraction (XRD, Rigaku D/max22500 XRD) with Cu Ka radiation (Rigaku Ltd, Japan). Magnetic properties were determined using vibrating sample magnetometry (VSM, Lake Shore Ltd) at room temperature.2.3 Preparation of MHNTs
MHNTs were synthesized according to our previous reported method. Crude HNTs were sieved and then dried at 100 °C in an oven for 12 h. HNTs weighing 0.5 g were briefly dispersed in 150 mL of deionized water by sonication for 15 min, and then 1.30 g of FeCl3·6H2O and 0.48 g of FeCl2·4H2O were added. The mixture was stirred for 10 min at 60 °C in N2 atmosphere. Subsequently, 10 mL of ammonia solution was added dropwise into the mixture solution. The addition of the base to the Fe2+/Fe3+ salt solution resulted in the immediate formation of black precipitates of MHNTs. Then, the resulting reaction mixture was aged for 4 h at 70 °C. The MHNTs were separated by an external magnetic field and washed for three times sequentially with water and ethanol. Finally, the MHNTs were dried in vacuum at 60 °C.2.4 Preparation of vinyl modified MHNTs
MPTES, as a silane coupling agent, was used to prepare the functionalized MHNTs (MHNTs@C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C). MHNTs weighing 250 mg were dispersed in 15 mL of anhydrous toluene in a three-necked flask. Then 3 mL of MPTES was added into the solution. The mixture was allowed to react for 24 h at 90 °C under constant stirring under an N2 atmosphere. After cooling to room temperature, the resultant was washed sequentially with water and ethanol. The MHNTs@CC was collected by an external magnetic field and dried in vacuum at 60 °C.
C). MHNTs weighing 250 mg were dispersed in 15 mL of anhydrous toluene in a three-necked flask. Then 3 mL of MPTES was added into the solution. The mixture was allowed to react for 24 h at 90 °C under constant stirring under an N2 atmosphere. After cooling to room temperature, the resultant was washed sequentially with water and ethanol. The MHNTs@CC was collected by an external magnetic field and dried in vacuum at 60 °C.
2.5 Preparation of MHNTs@BSA-MIP and MHNTs@NIP
MHNTs@BSA-MIP was obtained by the following method: 25.0 mg of BSA was dissolved in 20.0 mL of 0.1 mol L−1 phosphate buffer (pH 6.98) in a three-necked round-bottomed flask, and then 84.8 mg of NIPAM and 21.5 mg of MAA as co-monomers were added. The resulting mixture was dispersed homogeneously and shook for 5 h in the dark at room temperature to allow self-assembly. Subsequently, 200 mg of MHNTs, 7.8 mg of BIS, 10 mg of APS, and 10 μL of TEMED were added to the self-assembly solution. The solution was deoxygenated by purging with nitrogen for 15 min. The reaction was allowed to proceed at 35 °C for 24 h under mechanical stirring. Afterward, the products were separated by an external magnetic field and then repeatedly rinsed with distilled water to remove unreacted monomers and crosslinker. Then, the template protein was eluted with SDS–acetic acid (10% w/v:10% v/v) solution repeatedly until BSA was no longer detected in the supernatant. Finally, the products were washed with distilled water until the solution was neutral, and then dried in vacuum at 60 °C.
MHNTs@NIP was prepared as control by using the same procedure under the same conditions but without the template protein.
2.6 Adsorption experiments
Rebinding equilibrium and adsorption kinetics tests were performed to evaluate the recognition properties of MHNTs@BSA-MIP toward the template protein in batch mode operations.For this purpose, 10 mg of MHNTs@BSA-MIP or MHNTs@NIP was immersed into a 10 mL solution of BSA in PBS. The binding experiments were conducted in an incubator shaker at 25 °C for 24 h. After magnetic separation, the concentration of BSA in the supernatant was measured by a UV-vis spectrophotometer at 278 nm detection wavelength. The amount of protein adsorbed on the imprinted magnetic polymers was calculated based on the difference in BSA concentration before and after adsorption. All tests were conducted in triplicate.
The experimental data are presented as the adsorption capacity per unit mass (g) of MHNTs@BSA-MIP and the adsorption equilibrium capacity Qe (mg g−1) is calculated as follows:
| Qe = (Co − Ce)V/m | (1) |
In the adsorption kinetics tests, 10 mg of MHNTs@BSA-MIP or MHNTs@NIP was added to 10 mL of BSA solution at an initial concentration of 200 mg L−1 and investigated at regular time intervals from 5–180 min at room temperature.
2.7 Selective adsorption experiments
The selectivity of MHNTs@BSA-MIP was evaluated at equilibrium adsorption condition using Lyz, OVA, and Try as competitive proteins. The competitive proteins were chosen according to their MW and pI. Both Lyz and Try have relatively smaller sizes than the BSA, and they have different pI values, i.e., 11.2 and 7.8, respectively. OVA have a smaller MW of 43 kDa and a similar pI of 4.7.MHNTs@BSA-MIP or MHNTs@NIP (10 mg) was added to the mixture containing all of the proteins at a concentration of 200 mg L−1 and then incubated for 5 h. Then, the supernatants and polymers were separated by an external magnetic field, and the concentrations of proteins in the supernatant were measured by UV-vis spectrometry.
In addition, imprinting factor (IF) and selectivity coefficient (SC) were used to evaluate the selectivity property of MHNTs@BSA-MIP and MHNTs@NIP toward the template protein and competitive proteins. The IF and SC were calculated using the following equations:
| IF = QMIP/QNIP | (2) |
| SC = IFTEM/IFCOM | (3) |
3 Results and discussion
3.1 Selection of monomers
In this work, MHNTs@BSA-MIP was prepared using NIPAM and MAA as co-monomers. The role of the monomer is to provide functional groups, which can form a complex with the template by covalent or non-covalent interactions. The strength of the interactions between template and monomer affects the affinity of MIP and determines the accuracy and selectivity of imprinted sites. The stronger the interactions is, the more stable the resultant complex prior to polymerization is, resulting in the better imprinting efficiency of the MIP, and therefore, correct selection of the functional monomers is of great importance. Given that BSA possesses more flexible conformational transitions in the imprinting process owing to its larger molecular size, hydrogen bonds between monomer NIPAM and BSA are too weak to form stable BSA–monomer complexes. To assist the imprinting of acid protein BSA (pI 4.9), an electrostatic interaction between monomer and BSA is considered, which is supposed to benefit the formation of imprinted sites in polymers. Therefore, MAA was chosen as additive monomer in BSA imprinting process owing to the strong electrostatic interactions of carboxyl groups toward amino groups of BSA. An appropriate mass of BIS was added to ensure the formation of stable protein–monomer complexes via multiple-point electrostatic interactions.3.2 Preparation of MHNTs@BSA-MIP
The process for preparing a new kind of MHNTs@BSA-MIP was illustrated in Fig. 1, which combines the surface imprinting technique and a two-step immobilized template strategy. First, MHNTs were obtained according to a modified self-assembly coprecipitation method based on HNTs, and the surface Si–O–Si groups of MHNTs can be further modified through covalent attachment. MPS hydrolysis in toluene and its reaction with the Si–O–Si groups at the surface of the prepared MHNTs were allowed to proceed to synthesize MHNTs@CC, whose surfaces comprise numerous vinyl groups. The addition of BIS as a cross-linker maintained the stability of the imprinting sites while facilitating easier handling and processing of the produced polymeric beads. Subsequently, BSA was selectively immobilized on the surface of the MHNTs@CC through polymerization. After the removal of BSA, a thin polymer layer was obtained with imprinted cavities complementary to BSA in shape, size, and functional group orientation.
 | ||
| Fig. 1 Schematic representation of preparing MHNTs@BSA-MIP. | ||
3.3 Characterization
 | ||
| Fig. 2 TEM images of HNTs (a), MHNTs (b), and MHNTs@BSA-MIP (c). | ||
C (b), and MHNTs@BSA-MIP (c) were shown in Fig. 3, which provided direct evidences for the synthetic process of MHNTs@BSA-MIP. As shown in Fig. 3a, the absorption peaks at 535 and 468 cm−1 belonged to the stretching of the Fe–O vibration for Fe3O4 nanoparticles, indicating the successful synthesis of MHNTs. The bands observed at 3695 and 3624 cm−1 denoted the stretching bands of hydroxyl group of HNTs. Interlayer water was indicated by the bending vibration at 1638 cm−1. The peaks at 3445 and 1633 cm−1 were attributed to the characteristic absorptions of O–H stretching and CC vibration of MHNTs, respectively. The characteristic peaks of Si–O–Si and deformation vibration of inner-surface hydroxyl groups for MHNTs (a), MHNTs@CC (b), and MHNTs@BSA-MIP (c) were observed at around 1030 and 910 cm−1, respectively. The peak at 1715 cm−1 for MHNTs@CC indicated the successful functionalization with vinyl groups. The asymmetric stretching vibration of C–H of –CH2 group at 2921 cm−1 showed that the imprinted polymer was successfully grafted on the surface of MHNTs@BSA-MIP.
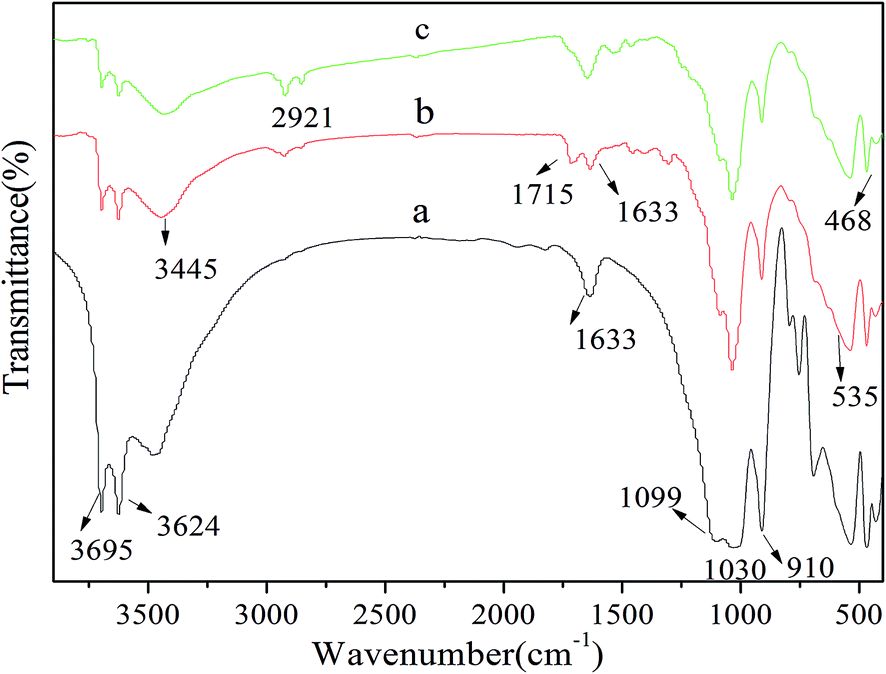 | ||
| Fig. 3 FT-IR spectra of the MHNTs (a), MHNTs@CC (b), and MHNTs@BSA-MIP (c). | ||
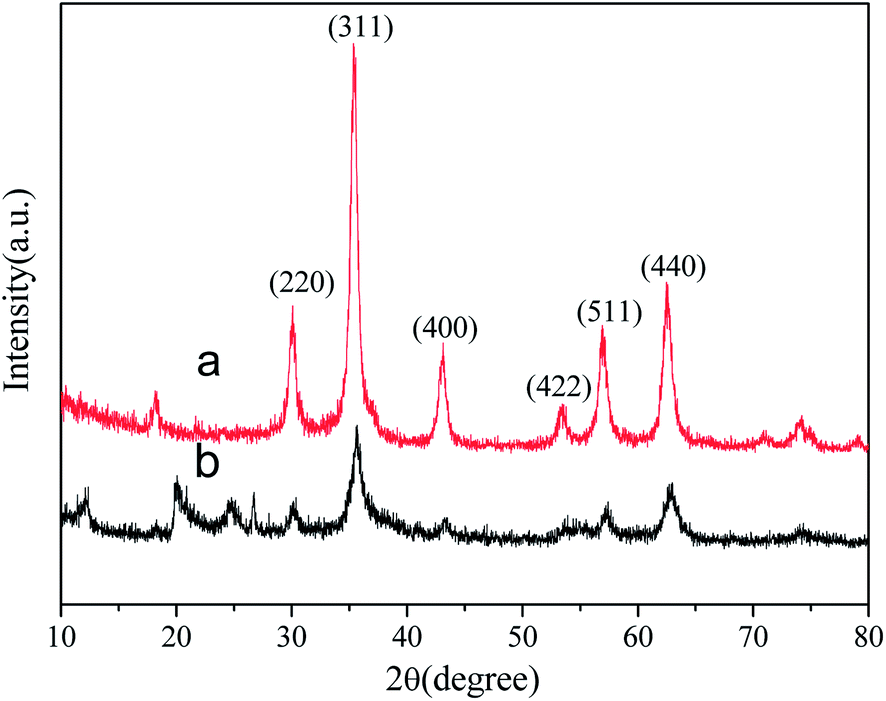 | ||
| Fig. 4 XRD patterns of Fe3O4 (a), and MHNTs@BSA-MIP (b). | ||
After precipitation polymerization with a crosslinking agent, the intensity of the XRD peaks decreased, which is attributed to the coating of the polymers on the MHNTs.
 | ||
| Fig. 5 The hysteresis loops of MHNTs (a), and MHNTs@BSA-MIP (b). The inset shows a photograph of the MHNTs@BSA-MIP dispersed in the water in the presence of an external magnetic field. | ||
3.4 Adsorption properties of MHNTs@BSA-MIP
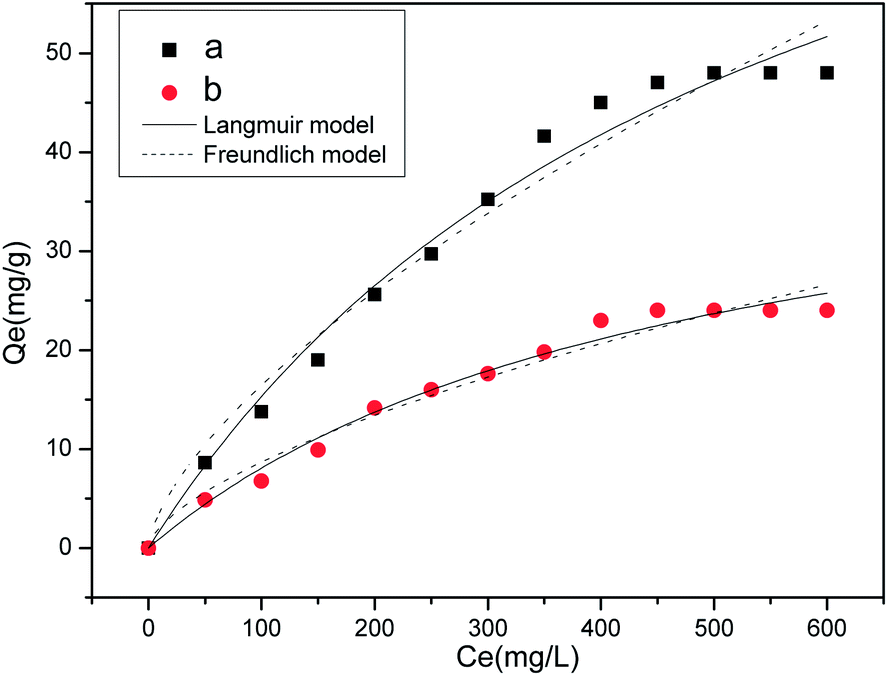 | ||
| Fig. 6 Equilibrium adsorption of MHNTs@BSA-MIP (a), and MHNTs@NIP (b). | ||
To evaluate the binding properties of MHNTs@BSA-MIP and MHNTs@NIP further, Langmuir and Freundlich models were selected to fit the experimental data according to the following equations:
 | (4) |
| Qe = KFCe1/n | (5) |
Table 1 summarized the fitted parameters Qm, KL, KF, 1/n, and R2 (correlation coefficient) and reveals that the Langmuir equation was found to better fit the isotherm data, with R2 > 0.98.
| Adsorbent | Langmuir model | Freundlich model | ||||
|---|---|---|---|---|---|---|
| KL | Qm | R | KF | 1/n | R | |
| MHNTs@BSA-MIP | 0.0018 | 98.34 | 0.9831 | 74.48 | 0.6557 | 0.9543 |
| MHNTs@NIP | 0.0021 | 45.81 | 0.9824 | 36.59 | 0.6246 | 0.9508 |
 | ||
| Fig. 7 Dynamic adsorption of MHNTs@BSA-MIP (a), and MHNTs@NIP (b). | ||
 | ||
| Fig. 8 Selectivity adsorption of MHNTs@BSA-MIP (a) and MHNTs@NIP(b). | ||
The selectivity of the MHNTs@BSA-MIP in terms of IF (α) and SC was compared with other work. The results were listed in Table 2. The MHNTs@BSA-MIP exhibited much higher absorptive capacity and IF for BSA than for the competitive proteins as shown in Table 2. On one hand, for the competitive proteins, no strong shape memory effects to enter into the imprinted cavities were found because they are much smaller than the imprinted cavities produced by BSA. On the other hand, they have different pI values from the template BSA. Thus, the microenvironment of binding process was unsuitable for the competitive proteins. The shape memory effect and multiple non-covalent interactions complementary to template protein in the form of electrostatic interaction, hydrogen bonding, and hydrophobic interaction play important roles in protein recognition in the MHNTs@BSA-MIP.
| Adsorbents | Supporter | IF | SC | Reference | |||||
|---|---|---|---|---|---|---|---|---|---|
| BSA | Lyz | OVA | Try | Lyz | OVA | Try | |||
| Fe3O4@SiO2@BSA-MIP | Fe3O4 | 1.70 | 1.27 | 1.12 | 1.34 | 1.52 | 17 | ||
| MHNTs@BSA-MIP | MHNTs | 2.27 | 1.08 | 1.11 | 1.30 | 2.10 | 2.05 | 1.75 | This work |
3.5 Regeneration and reproducibility
Regeneration and reusability is one of the most important properties for the application of MHNTs@BSA-MIP. The adsorption–desorption cycles were repeated for several times for the same batch of MIP to verify their regeneration and reusability. The MHNTs@BSA-MIP after BSA absorption was washed with an SDS–acetic acid (10% w/v:10% v/v) solution to remove BSA and applied to rebind BSA. As shown in Fig. 9, the adsorption capacity for BSA showed only a slight decrease after continuous adsorption and desorption processes, whereas the adsorption capacity of MHNTs@NIP toward BSA remained unchanged. This finding could be attributed to changes in special recognition sites that are sterically complementary to BSA caused by repeated washing. However, given that the MHNTs@NIP had no special recognition sites, the affinity of MHNTs@NIP was nonspecific, and the effect of washing was negligible. The data confirmed that the MHNTs@BSA-MIP has an outstanding reusability.
 | ||
| Fig. 9 The stability and regeneration of MHNTs@BSA-MIP (a) and MHNTs@NIP (b). | ||
4 Conclusion
In this work, a facile procedure for preparation of MHNTs@BSA-MIP was developed by combining a surface imprinting technique and magnetic separation technology for selective recognition of BSA. Attachment of Fe3O4 on the surface of HNTs was achieved by coprecipitation method, which occurred in the presence of two ferric salts and pure HNTs. The MHNTs@BSA-MIP was prepared using modified MHNTs as support. The MHNTs@BSA-MIP demonstrated saturation magnetization, rapid dynamic adsorption, high adsorption ability, and a higher reusability for adsorption of the template protein than the MHNTs@NIP. Separation was easily done by using an external magnetic field. The MHNTs@BSA-MIP exhibited potential application in protein separation, solid-phase extraction, chromatographic separation, drug delivery, and medical diagnostics.More detailed investigation on enhancing the imprinting efficiency in terms of IF and specific rebinding capacity should be conducted. Follow-up work and appropriate measures should be performed to adjust the thickness and composition of MIP. For example, the amount of functional monomers with stronger specific adsorption for the template molecule may be increased or varied to improve IF.
Acknowledgements
The authors greatly acknowledge the financial support from National Natural Science Foundation of China (21276283).References
- E. Ueda, P. Gout and L. Morganti, J. Chromatogr. A, 2003, 988, 1–23 CrossRef CAS.
- J. A. Asenjo and B. A. Andrews, J. Chromatogr. A, 2012, 1238, 1–10 CrossRef CAS PubMed.
- P. G. Righetti, R. Sebastiano and A. Citterio, Proteomics, 2013, 13, 325–340 CrossRef CAS PubMed.
- X. Jia, M. Xu, Y. Wang, D. Ran, S. Yang and M. Zhang, Analyst, 2013, 138, 651–658 RSC.
- A. Poma, A. Guerreiro, S. Caygill, E. Moczko and S. Piletsky, RSC Adv., 2014, 4, 4203–4206 RSC.
- J. Pan, W. Zhu, X. Dai, X. Yan, M. Gan, L. Li, H. Hang and Y. Yan, RSC Adv., 2014, 4, 4435–4443 RSC.
- S. Zhong, Y. Kong, L. Zhou, C. Zhou, X. Zhang and Y. Wang, J. Chromatogr. B, 2014, 945–946, 39–45 CrossRef CAS PubMed.
- E. Verheyen, J. P. Schillemans, M. van Wijk, M.-A. Demeniex, W. E. Hennink and C. F. van Nostrum, Biomaterials, 2011, 32, 3008–3020 CrossRef CAS PubMed.
- J. Liu, K. Yang, Q. Deng, Q. Li, L. Zhang, Z. Liang and Y. Zhang, Chem. Commun., 2011, 47, 3969–3971 RSC.
- H. Chen, J. Kong, D. Yuan and G. Fu, Biosens. Bioelectron., 2014, 53, 5–11 CrossRef CAS PubMed.
- Y.-Q. Yang, X.-W. He, Y.-Z. Wang, W.-Y. Li and Y.-K. Zhang, Biosens. Bioelectron., 2014, 54, 266–272 CrossRef CAS PubMed.
- M. E. Çorman, C. Armutcu, L. Uzun, R. Say and A. Denizli, Colloids Surf., B, 2014, 123, 831–837 CrossRef PubMed.
- D.-Y. Li, Y.-P. Qin, H.-Y. Li, X.-W. He, W.-Y. Li and Y.-K. Zhang, Biosens. Bioelectron., 2015, 66, 224–230 CrossRef CAS PubMed.
- G. Pan, Q. Guo, C. Cao, H. Yang and B. Li, Soft Matter, 2013, 9, 3840–3850 RSC.
- M. Zhang, Y. Wang, X. Jia, M. He, M. Xu, S. Yang and C. Zhang, Talanta, 2014, 120, 376–385 CrossRef CAS PubMed.
- K. Zhao, B. Lin, W. Cui, L. Feng, T. Chen and J. Wei, Talanta, 2014, 121, 256–262 CrossRef CAS PubMed.
- X. Li, B. Zhang, W. Li, X. Lei, X. Fan, L. Tian, H. Zhang and Q. Zhang, Biosens. Bioelectron., 2014, 51, 261–267 CrossRef CAS PubMed.
- R. Gao, X. Mu, Y. Hao, L. Zhang, J. Zhang and Y. Tang, J. Mater. Chem. B, 2014, 2, 1733–1741 RSC.
- H. Guo, D. Yuan and G. Fu, J. Colloid Interface Sci., 2015, 440, 53–59 CrossRef CAS PubMed.
- H. He, G. Fu, Y. Wang, Z. Chai, Y. Jiang and Z. Chen, Biosens. Bioelectron., 2010, 26, 760–765 CrossRef CAS PubMed.
- R. Ouyang, J. Lei and H. Ju, Chem. Commun., 2008, 5761–5763 RSC.
- G. Fu, H. He, Z. Chai, H. Chen, J. Kong, Y. Wang and Y. Jiang, Anal. Chem., 2011, 83, 1431–1436 CrossRef CAS PubMed.
- Z. Lin, Z. Xia, J. Zheng, D. Zheng, L. Zhang, H. Yang and G. Chen, J. Mater. Chem., 2012, 22, 17914–17922 RSC.
- X. Hu, L. Xie, J. Guo, H. Li, X. Jiang, Y. Zhang and S. Shi, Food Chem., 2015, 179, 206–212 CrossRef CAS PubMed.
- D. Zhu, Z. Chen, K. Zhao, B. Kan, H. Li, X. Zhang, B. Lin and L. Zhang, RSC Adv., 2015, 5, 26977–26984 RSC.
- L. Yuan, L. Jiang, T. Hui, L. Jie, X. Bingbin, Y. Feng and L. Yingchun, Sens. Actuators, B, 2015, 206, 647–652 CrossRef CAS PubMed.
- H. Duan, L. Li, X. Wang, Y. Wang, J. Li and C. Luo, RSC Adv., 2015, 5, 18850–18857 RSC.
- R. Gao, Y. Hao, X. Cui, L. Zhang, D. Liu and Y. Tang, J. Alloys Compd., 2015, 637, 461–465 CrossRef CAS PubMed.
- S. Xu, H. Lu, L. Chen and X. Wang, RSC Adv., 2014, 4, 45266–45274 RSC.
- H.-J. Chen, Z.-H. Zhang, L.-J. Luo and S.-Z. Yao, Sens. Actuators, B, 2012, 163, 76–83 CrossRef CAS PubMed.
- J. Huang, X. Xing, X. Zhang, X. He, Q. Lin, W. Lian and H. Zhu, Food Res. Int., 2011, 44, 276–281 CrossRef CAS PubMed.
- W. O. Yah, H. Xu, H. Soejima, W. Ma, Y. Lvov and A. Takahara, J. Am. Chem. Soc., 2012, 134, 12134–12137 CrossRef CAS PubMed.
- W. O. Yah, A. Takahara and Y. M. Lvov, J. Am. Chem. Soc., 2012, 134, 1853–1859 CrossRef CAS PubMed.
- C. Zhou, H. Li, H. Zhou, H. Wang, P. Yang and S. Zhong, J. Sep. Sci., 2015, 38, 1365–1371 CrossRef CAS PubMed.
- J. Pan, B. Wang, J. Dai, X. Dai, H. Hang, H. Ou and Y. Yan, J. Mater. Chem., 2012, 22, 3360 RSC.
- J. Dai, X. Wei, Z. Cao, Z. Zhou, P. Yu, J. Pan, T. Zou, C. Li and Y. Yan, RSC Adv., 2014, 4, 7967 RSC.
- J. Pan, H. Yao, L. Xu, H. Ou, P. Huo, X. Li and Y. Yan, J. Phys. Chem., 2011, 115, 5440–5449 CAS.
- S. Zhong, C. Zhou, X. Zhang, H. Zhou, H. Li, X. Zhu and Y. Wang, J. Hazard. Mater., 2014, 276, 58–65 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |