
Luminescence properties and crystal structure of α′-Sr2Si3x/4O2Nx:Eu2+ phosphors with different concentrations of N3− ions
Abstract
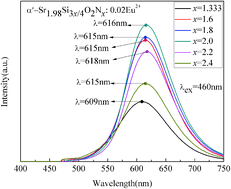
A series of disordered α′-Sr2Si3x/4O2Nx:Eu2+ (1.333 ≤ x ≤ 2.4) phosphors were synthesized by the conventional solid state reaction method. The disordered α′-Sr2Si3x/4O2Nx:Eu2+ (α′-SSON:Eu2+) phosphors have two distinct activation centers: Eu(I) and Eu(II). With the increase of N concentration, both the luminescence intensity and the dominant peak wavelength (DPWs, which is about 490 nm) of the Eu(I) site were extraordinarily unchanged. In comparison with the yellow emissions (∼580 nm) of the Eu(II) site of the disordered α′-Sr2SiO4:Eu2+, the DPWs of Eu(II) emissions were at red spectral regions (609–618 nm), which depends on the amount of N3−. The PL intensity of the Eu(II) emission band increased first and then decreased, and reached a maximum at x = 2. The disordered α′-SSON is a substitutional solid solution. Compared with the disordered α′-Sr2SiO4, all the lattice constants of disordered α′-SSON became smaller which led to the decrease of the cell volume. The peaks of the Si–N and Sr–N bond could be observed in FT-IR spectra. The Si–(N/O)4 tetrahedrons transformed from Si–O4, Si–NO3, and Si–N2O2 into Si–N3O with the increase of N content. The bond lengths of Si–N and Sr–(N/O) were within the normal ranges compared with other silicon-based oxynitrides. The Si–O bond lengths became shorter due to the extrusion effects of longer Si–N bonds. Both of the average bond lengths of Sr1–(N/O) and Sr2–(N/O) in disordered α′-SSON became longer than that of disordered α′-Sr2SiO4. Due to the red emission and high photoluminescence intensity of the disordered α′-Sr2Si3x/4O2Nx:Eu2+ (1.333 ≤ x ≤ 2.4), we anticipate that these materials can be used as red phosphors in white light emitting diodes.
 Please wait while we load your content...
Please wait while we load your content...

