
Exocyclic self-assembly behavior of carboxylic acid and lariat ether macrocyclic hosts: regulation by pendent arm†
Rong Guo,
Wei Wang,
Weiping Yang,
Yuanyin Chen and
Shuling Gong*
College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, 430072, P. R. China. E-mail: gongsl@whu.edu.cn; Fax: +86 27 68754067; Tel: +86 27 68752701
First published on 31st July 2015
Abstract
Interaction of 1,4-dicarboxybenzene (PTA) with N,N′-disubstituented dibenzo-diaza-18-crown-6-ethers (I, II, III, IV) bearing flexible side chain arms afforded the exocyclic supramolecular complexes of compositions 1 (I·PTA), 2 (II·PTA·0.2H2O), 3 (III·PTA) and 4 (IV·PTA), while with N-substituented dibenzo-aza-18-crown-6-ethers (V, VI, VII) afforded the endo-coordinated H2O salts 5 [3(V·H2O)·(3PTA·7H2O)], 6 [(VI·H2O)·(PTA·3H2O)] and 7 [2(VII·H2O)·(PTA·11H2O)], whose structures were determined by a single crystal X-ray method. These products were also obtained by the same synthetic conditions. Single crystal X-ray structural investigations on these solids confer: P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space symmetry for compound III and complexes 3, 5 and 7; C2/c space symmetry for the complexes 1, 2 and 4; P21/n space symmetry for compound I; P212121 for complex 6. The macrocyclic entities in compounds I, III and complexes 1, 2, 3, 4 with the ‘chair-like’ conformation of the crown ether ring and the extended arrangement of the pendent arms, adopt the structure as ‘out–out’ cyclic amines. But the macrocyclic entities in salts 5 and 6 adopt the C-shape conformation. The side chain arm may introduce minor crystal structure disparity, particularly 3D packing types in the exocyclic complexes 1, 2, 3 and 4, with the arrangement of the pendent arms in protonated mono-sidearm crown ethers. Moreover, each complex with distinct ionic states, indicates that the proton transfer depends also upon the solid state environment.
space symmetry for compound III and complexes 3, 5 and 7; C2/c space symmetry for the complexes 1, 2 and 4; P21/n space symmetry for compound I; P212121 for complex 6. The macrocyclic entities in compounds I, III and complexes 1, 2, 3, 4 with the ‘chair-like’ conformation of the crown ether ring and the extended arrangement of the pendent arms, adopt the structure as ‘out–out’ cyclic amines. But the macrocyclic entities in salts 5 and 6 adopt the C-shape conformation. The side chain arm may introduce minor crystal structure disparity, particularly 3D packing types in the exocyclic complexes 1, 2, 3 and 4, with the arrangement of the pendent arms in protonated mono-sidearm crown ethers. Moreover, each complex with distinct ionic states, indicates that the proton transfer depends also upon the solid state environment.
Introduction
Crystal engineering is a powerful technique to reveal the role of various noncovalent interactions assembling molecules as crystals with desired structures and specific chemical/physical properties.1 However, the main impediments of designing and modifying noncovalently bound supramolecular structures are: (i) the multiplicity of possible orientations of the molecules in crystals; (ii) the inaccuracies in estimating energies; and (iii) the entanglement of thermodynamic and kinetic contributions to crystal growth.2 It has been recognized that to limit the possible arrangement of the molecules in the solid state with respect to one another has been considered one of the most rational approaches to gain control over the arrangement of molecules in space, by incorporation of a small number of functional groups.2,3 The supramolecular synthon is a powerful concept for crystal engineering in the design of zero- to three-dimensional (0D to 3D) architectures.4 Among the synthons, with a robust and anisotropic nature, hydrogen-bonding (HB) synthons are quite useful in designing low-dimensional supramolecular motifs.5 Recently, HB synthons based on carboxylic acid–amine complexes have drawn much attention from the crystal engineering community.6 These groups are useful in various applications such as chiral synthesis7 and separation,8 supramolecular low-dimensional assemblies,3,9 and topological supramolecular chirality.6c,10 Supramolecular building blocks based on amines and carboxylic acids5c,11 offer good chemical diversity and provide a useful structural handle in their preference for 1D geometries. Besides, organic salts12 and co-crystals12a,13 could be distinguished by the protonation–deprotonation behavior in carboxylic acid–amine complexes.12a,14 Both ionic hydrogen bonds in organic salts and hydrogen bonds in co-crystals have been investigated extensively. Furthermore, there are many works15 proving that the competition for hydrogen bonds between different functional groups in forming particular intermolecular interactions definitely affects the predictability of a particular supramolecular synthon, influencing the design of supramolecular structures.Aza-crown ethers16 are a well-known family of macrocyclic compounds17 with important uses as good coordination agents for a wide range of metals and cations. The anion complexation properties are similar to those in certain biological system due to their nitrogen lone pairs and N–H moieties. The uses of aza-crown ethers to form complexes with organic acids are also quite common.18 As we know, unlike oxygen-bearing crown ethers, aza-bearing analogues have a tendency to show a repulsive interaction between adjacent nitrogen donors; the interaction leads to a trans (or anti) torsion arrangement, akin to the sulfur-containing macrocycles.16a Lee19 concluded that the exocyclic coordination behavior in sulfur containing macrocycles may result from the repulsive interaction between adjacent sulfur donors. The strategy for macrocyclic ligand-based crystal engineering is to tune the structural and electronic features of a given macrocycle according to the guest. This should be practicable for the preparation of new molecular networks and materials based on aza-crown ethers by controlling the orientation of the lone pair electrons on N atoms. The ‘out–in’ designation (Scheme 1), referring to the orientation of the nitrogen lone pairs relative to the interior or external in out–in bicyclic amines and cryptands proposed by Simmons,20 could also be applied to aza-crown ethers. The interactions by which guests attach to the aza-crown ethers are: head-to-face (HF),16b,16c,17a,17c,17d,21 head-to-edge (HE)18 and head-to-head (HH); of which type (HE) and type (HF) have been experimentally verified by supramolecular networks of macrocycles based on exo and endo-coordination. However, type (HH) in the diaza-18-crown-6 exo-coordination compounds has not been fully explored yet. Fonari’s studies18a,22 on lariat crowns and diaza-crown ethers showed both type (HE) and type (HF) with a flexible macrocycle and rigid pendent. In contrast with an aliphatic crown ether, the stability constants of benzo-crown ethers are known to be lower, due to their higher rigidity and the lower basicity of the oxygens caused by their conjugation with the aromatic ring.17b For this property, the conformation of the dibenzo-azacrown ether may be akin to the semi-rigid dibenzo-crown ether: C-shape16b,23 and S-shape.24 Disparities between them could be caused by the various orientations of the lone pair electrons of N atoms and substituent groups. The relationship between the macrocyclic conformations and the orientations of the lone pair electrons in N atoms are shown in Scheme 1.25 According to this, the steric hindrance of the substituent group on the N atom would have an influence on the orientation of the lone pair electrons for dibenzo-aza-18-crown-6 ethers. To investigate the influence of side chains on the host–guest chemistry based on azacrown ethers, we have chosen terephthalic acid (PTA) with a linear functional topology and a series of semi-rigid macrocycles, N,N′-disubstituented dibenzo-diaza-18-crown-6s (DBDA18C6) and N-substituented dibenzo-aza-18-crown-6s (DBA18C6), bearing flexible substituent groups as the host (Scheme 2), resulting in applications such as molecular (or ion) sensing and exchange. The exocyclic head-to-edge or head-to-head arrangement of PTA as the robust supramolecular motif may be preserved in the crystals.
 | ||
| Scheme 1 The “out–in” designation refers to the orientation of the nitrogen lone pairs in diaza-crown ethers: out–out (left), in–in (right), conformation with axial positions of lone pairs (middle), and possible motifs of host–guest. | ||
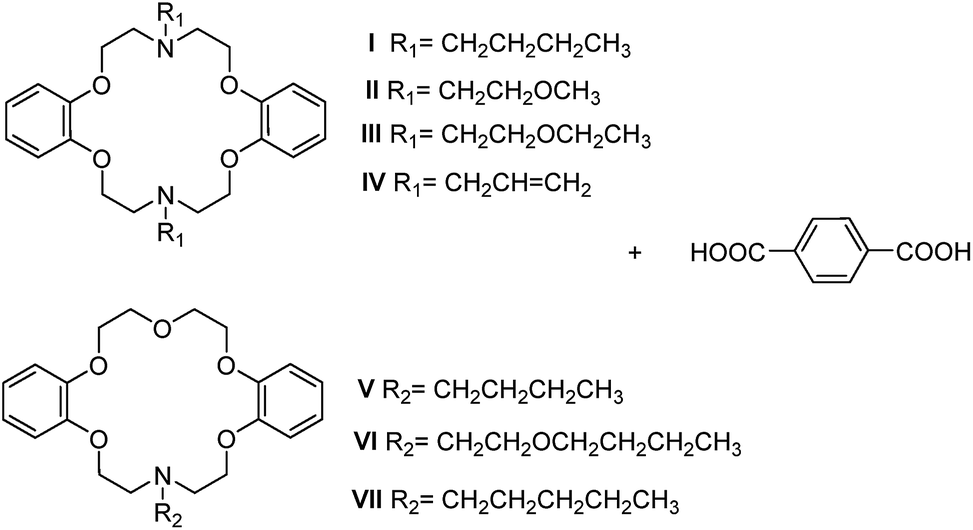 | ||
| Scheme 2 Target 1,4-dicarboxybenzene and lariat ethers. | ||
Whether proton transfer has occurred from acid to base, as Childs12b noted, depends upon both the ΔpKa value (pKa of base − pKa of acid) and the crystalline environment. According to the studies by Nangia,26 the transfer of a proton is uncertain in the range 0 < ΔpKa < 3.75, but above 3.75 it is salt and below 0 it forms a neutral complex. For PTA pKa1 = 3.49, 25 °C and pKa2 = 4.46, 25 °C; and for azacrown ether I, II, III, IV, V, VI, VI, pKa = 7–8, 25 °C (predicted by SciFinder); this would be complicated with salts or co-crystals. But in a solid, the nature (co-crystal/salt) of the complex is generally determined by the location of the H atom and the bond lengths of two C–O bonds in the carboxylic group. It has been elucidated that the two C–O bond distances are comparable (ΔDC–O < 0.03) for a carboxylate ion, whereas they are distinct (ΔDC–O > 0.08) for a neutral carboxyl group.12b IR spectra have been recorded, and looking for the disappearance of the bands due to COOH and appearance of the bands due to COO− in the case of deprotonation and appearance of the bands due to NH+, is helpful to clarify whether protonation of amines occurs or not.
For the first time, the structures of compounds (I, III) and the complexes of PTA with di-sidearm DBDA18C6s and mono-sidearm DBA18C6s in solid were isolated and characterized by X-ray single-crystal diffraction. In this paper, by choosing a semi-rigid macrocycle and similar side chain groups, we have shown that the pendent arms affect the supramolecular structure of complexes 1, 2, 3, 4, 5, 6 and 7 in same crystallization medium and the different protonation–deprotonation behaviors by using X-ray single crystal diffraction and FT-IR spectra. Also in the discussion about the packing modes of the molecular crystals, it is important to understand non-covalent interactions such as the N–H⋯O, O–H⋯N, C–H⋯O hydrogen bondings27 and C–H⋯π28 or π⋯π stacking interactions,29 which play a critical role in controlling the packing modes of molecular crystals.
Results and discussion
Crystal structures of compounds I and III
Free macrocycle I crystallizes in the monoclinic space group P21/n with half a molecule in the asymmetric unit. The crystal of macrocycle III is in a triclinic cell, and structure solution was performed with the space group P; the asymmetric unit contains half of a DBDA18C6 (III) molecule. The oxygen atoms in compounds I and III are arranged in an endo-dentate mode typical for most crowns.17b The 18-crown-6 molecule displays a ‘chair-like’ conformation in I and III (Fig. 1); two methylene groups on opposite sides of the ring are in the anti conformation and are turned inward to fill the macrocyclic central void. The two nitrogen atoms extend out in a trans-fashion with the pendent arms in the axial position, apt to the ‘out–out’ conformation (Scheme 1). Obviously, Fig. 1 shows lariat ethers I and III both in the extended conformation. These structures have not been reported yet, but show both differences from the aliphatic azacrown ethers and dibenzo-crown ethers. As we know, the benzene may be present as a subcyclic unit that contributes no heteroatom to the macrocycle but does not otherwise alter the essential (O–C–C)n crown ether framework. This has obvious effects on the macrocyclic structure. Firstly, replacement of OCH2–CH2OCH2CH2O by OCH2C–CH–CCH2O rigidifies the macrocycle. Secondly, each replacement results in the loss of a donor group relative to diethyleneoxy.17b So, the semi-rigid feature of the macrocycle and the flexibility of the pendent arms can be explained for the structures of I and III in the solid.
 | ||
| Fig. 1 (a) X-Ray crystal structure of compound I; (b) X-ray crystal structure of compound III; (c) and (d) fragment of crystal packing in I and III showing the association of the molecules in a ladder via C–H⋯π and C–H⋯O interactions. | ||
In compound I, along the b axis two uniform macrocycle stacks interacted via C–H⋯π interactions (C7–H7B⋯Cg(crown): H7B⋯Cg 2.65 Å, C7⋯Cg 3.468(3) Å, C7–H7B⋯Cg 140°) and C–H⋯O hydrogen bonding interactions (C8–H8A⋯O2: H⋯O = 2.668 Å, C⋯O = 3.578 Å, C–H⋯O = 152°), forming a ladder-like structure. Similar packing stacks along the c axis in compound III (C10–H10B⋯Cg(crown): H10B⋯Cg = 2.79 Å, C10⋯Cg = 3.576(2) Å, C10–H10B⋯Cg = 138°; C9–H9B⋯O1: H⋯O = 2.737 Å, C⋯O = 3.659 Å, C–H⋯O = 159°). Generally the intermolecular interactions of C–H⋯O(OCH2) hydrogen bonding based on the methoxy groups in the crown systems results in a stepwise packing structure. In the structures of compounds I and III, the C–H⋯π interactions should also be beneficial to the packing, as they are even more available.
Crystal structure of complex 1 (I·PTA)
Single crystals of 1 were prepared in a monoclinic cell and structure solution was performed in the space group C2/c. The asymmetric unit of 1 was found to contain a half moiety of PTA acid and DBDA18C6 (I). The structure is stabilized via O1–H1⋯N1 hydrogen bonds connecting the para-hydroxyl group of PTA and the DBDA18C6 moiety molecule, respectively (Fig. 2a). It is idiosyncratic for the PTA to attach with crown in the head-to-head arrangement rather than head-to-face. Hydrogen bonding interactions [O1–H1⋯N1 = 1.77(2) Å; O1–H1⋯N1 = 172(5)°] involving the N of the DBDA18C6 and OH of the PTA moieties lead to the formation of a 1D motif A (Scheme 1); the 1D linear ribbons are further packed in parallel fashion along the b axis (Fig. 2b), forming a staircase-like layer. Two layers of crystal packing of 1 are demonstrated in Fig. 2b with the C–H⋯π interactions [aromatic CH/π hydrogen bond C7–H7⋯Cg(crown): H7⋯Cg = 2.93 Å, C7⋯Cg = 3.751(3) Å, angle C7–H7⋯Cg = 146°. In addition, C18–H18D⋯Cg(PTA) hydrogen bond: H18D⋯Cg = 3.24 Å, C18⋯Cg = 3.815(4) Å, angle C18–H18D⋯Cg = 119°]. | ||
| Fig. 2 Illustration of the single crystal structures of complexes 1, 2, 3, 4: (a), (c), (e), (g) hydrogen bonding interactions forming 1D motif; (b), (d), (h): parallel packing of the 1D networks along the b axis through C–H⋯O hydrogen bondings of complexes 1, 2, 4, respectively (broken line). (f) Parallel packing of the 1D networks along the a axis through C–H⋯O hydrogen bondings of complex 3 (broken line). | ||
Crystal structure of complex 2 (II·PTA·0.2H2O)
The complex 2 crystallized in the monoclinic space group C2/c. The asymmetric portion of the unit cell contains 0.5 crown ether: 0.5 PTA. Hydrogen bonding interactions [O1–H1A⋯N1 = 1.74(2) Å; O1–H1A⋯N1 = 164(5)°] involving the OH of the PTA and N of the crown moieties lead to the formation of 1D motif A (Scheme 1); along the b axis, the 1D ribbons are further packed in a parallel fashion via the C–H⋯O hydrogen bonding interactions (Fig. 2c). Lattice occluded water molecules are also observed within the bilayer structure stabilized by the water–carboxylate hydrogen bonding interactions (Fig. 3B and b). The water molecules bridge the two PTA molecules via O6–H6A⋯O2 hydrogen bonding [O6–H6A⋯O2 = 2.16 Å; O6–H6A⋯O2 = 161.9°] with 21 symmetric arrangement along the c axis (Fig. 3B). It can also be noted here that weak interactions such as C7–H7⋯π(aromatic CH/π hydrogen bond: H7⋯Cg = 2.81 Å, C7⋯Cg = 3.626(3) Å, C7–H7⋯Cg = 144°) and C17–H17A⋯π(H17A⋯Cg = 2.87 Å, C17⋯Cg = 3.532(9) Å, C17–H17A⋯Cg = 126°) also play a role in shaping the 3D supramolecular architectures in the solid, similar to the complex 1. The TGA measurement (ESI: Table S11†) of complex 2 shows a weight loss of 0.60% in the temperature range 175–190 °C, which corresponds to the loss of lattice water molecules (theoretical value of 0.56%). | ||
| Fig. 3 (A) The three-dimensional network of complex 1. (B) The three-dimensional network of complex 2. (C) The three-dimensional network of complex 3. (D) The three-dimensional network of complex 4. (a)–(d) The details of the hydrogen bonding interactions between the layers for each complex (macrocycles and PTA molecules in same sheet are shown with the same color in the 3d structure). | ||
Crystal structure of complex 3 (III·PTA)
Complex 3 crystallized in the centrosymmetric triclinic space group P. The asymmetric unit is composed of half of the DBDA18C6 moiety and the PTA moiety. As elucidated in Fig. 4a, hydrogen bonding interactions [O1–H1B⋯N1 = 1.71(6) Å; O1–H1B⋯N1 = 176(5)°] involving the OH of the PTA and N of the crown moieties lead to the formation of 1D motif A (Scheme 1). Along the a axis, the 1D linear ribbons are further packed parallel by C–H⋯O hydrogen bonding interactions (Fig. 2e), which form the ‘inclined staircase’ step. Meanwhile, the mutual arrangement of the layers is attributed to a ‘staircase’ packing motif where adjacent ‘staircases’ are arranged in parallel by the π⋯π interactions (Cg⋯Cg = 4.12 Å, perpendicular distance 3.56 Å) (ESI: Table S10b†) between the benzenes of macrocycles in different layers (Fig. 3C). Furthermore, the interlayer C(pendent arm)–H⋯π(PTA) hydrogen bonding interactions (C18–H18C⋯π: H18C⋯Cg = 3.11 Å, C18⋯Cg = 3.85 Å, C18–H18C⋯Cg = 133°) can also be found in complex 3.
 | ||
| Fig. 4 Illustration of the single crystal structures of salts 5, 6 and 7: (a)–(c) conformation of mono-sidearm crown ethers in each salt; (d)–(f) the structure of crown ether sheet in each salt, respectively. (g)–(i) The layer structures of each salt (in 3d structure: magenta: crown ether; yellow: PTA; and red space-filling: the water in the cavity, hydrogen atoms are omitted in the sheet and the view direction of the 3D layered structure: (g) [−1, 1, 0]; (h) [0, 1, 0]; (i) [−1, 0, 1]). | ||
Crystal structure of complex 4 (IV·PTA)
The complex 4 crystallized in the monoclinic space group C2/c. The asymmetric unit is composed of half of the DBDA18C6 moiety and the PTA moiety. Hydrogen bonding interactions [O1–H1⋯N1 = 1.75(4) Å; O1–H1⋯N1 = 177(4)°] involving the OH of the PTA and N of the crown moieties lead to the formation of 1D motif A (Scheme 1) suggesting a ribbon structure that is sandwiched between DBDA18C6·PTA units; the 1D linear ribbons are further packed in a parallel fashion by the C–H⋯O hydrogen bonding interactions (Fig. 2e and f). Interestingly, the 1-D linear ribbons build staircase-like columns via C–H⋯O(2.41 Å, 2.56 Å) hydrogen bond interactions between PTA and the side arm of the DBDA18C6, as well as weak C–H⋯O(2.62 Å, 2.66 Å) interactions between two parallel macrocycles. Layers are further packed by the C–H⋯π interactions [aromatic CH/π hydrogen bond C7–H7⋯Cg(crown): H7⋯Cg = 2.73 Å, C7⋯Cg = 3.542(3) Å, angle C7–H7⋯Cg = 144°. And C17–H17B⋯Cg(PTA) hydrogen bond: H17B⋯Cg = 3.27 Å, C17⋯Cg = 4.086(4) Å, angle C17–H17B⋯Cg = 145°] (Fig. 3D); the staircases are arranged in a herringbone-type packing.Crystal structures of complex 5
The complex 5 [3(V·H2O)·(3PTA·7H2O)] crystallizes in the triclinic space group P, and has a layered structure containing a water–crown ether inclusion. These waters are incorporated within the cavity of the protonated 18-crown-6 by N–H⋯O and O–H⋯O hydrogen bonds as shown in Fig. 4. The asymmetric unit is comprised of three macrocyclic cations, three PTA anions, and ten water molecules. These water–crown ether inclusions are aggregated via C–H⋯π interactions, resulting in a chain. These chains are further connected into a sheet through π⋯π interactions (Cg⋯Cg = 4.57 Å, perpendicular distance 3.64 Å). Seated in the interlayer are seven water molecules and three PTA anions complexes that play a vital role in controlling the topology of the structures and also function in charge balancing. Additionally, hydrated anion layers are connected with crown ether layers via C–H(crown ether)⋯O(water) hydrogen bonds [H17B⋯O18 = 2.59 Å, C17B⋯O18 = 3.577(5) Å, C17–H17B⋯O18 = 173°]. The macrocycles adopt a distorted C-shape conformation, with the pendent arm behind the macrocyclic back in an axial position (Fig. 4g).
Crystal structures of complex 6
The complex 6 [(VI·H2O)·(PTA·3H2O)], crystallized in the orthorhombic space group p212121 with one crown ether molecule, one PTA and ten H2O molecules contained in the asymmetric unit, has a layered structure. These layers of water–crown ether inclusions are less bent than in salt 5. These waters are incorporated within the cavity of the protonated 18-crown-6 by N–H⋯O and O–H⋯O hydrogen bonds as shown in Fig. 4. Two macrocycles form a dimer via C–H⋯π interactions [C18A–H18B⋯π(phenylate): H⋯Cg = 2.59 Å, H-Perp = 2.56 Å, C–H⋯Cg = 144 Å; C18B–H18C⋯π(phenylate): H⋯Cg = 2.76 Å, H-Perp = 2.70 Å, C–H⋯Cg = 142 Å]. These crown ether dimers are further packed into a sheet via C–H⋯π and C–H⋯O interactions. The interlayer space is filled with planar solvated PTA anion complexes as shown in Fig. 4h. This is essential so that the overall charge balance is maintained. Meanwhile, the crown ether dimers are sandwiched between two solvated PTA anion layers via C–H⋯π and C–H⋯O interactions. Interestingly, the protonated macrocycle adopts the typical C-shape conformation similar to salt 5, but with the pendent arm to the front of the macrocycle and with a folded arrangement.Crystal structures of complex 7
The complex 7 [2(VII·H2O)·(PTA·11H2O)] with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, obtained from a 2:1 ratio of PTA and DBA18C6 (VII) from water/acetone, crystallizes in P space group with two DBA18C6 (VII) and one PTA moiety in the asymmetric unit, and has a layered structure containing a water–crown ether inclusion. These waters are incorporated within the cavity of the protonated 18-crown-6 by N–H⋯O and O–H⋯O hydrogen bonds. These inclusions are aggregated via C–H⋯π interactions (aromatic interaction, T-shape), resulting in a 1D chain (Fig. 4f). The chains are further connected through the C–H⋯O, C–H⋯π and π⋯π interactions into a sheet (ESI: Tables S9 and S10†). In this 2:1 structure, both of the carboxyl groups of PTA are deprotonated. Hydrated anion layers are connected with cation crown ether layers via O–H(water)⋯O(crown), C–H(crown ether)⋯O(water), and C–H(crown ether)⋯O(PTA) hydrogen bonds (Fig. 4i). The conformation of the macrocycle and the arrangement of the pendent arm are similar to complex 5.
:1 ratio, obtained from a 2:1 ratio of PTA and DBA18C6 (VII) from water/acetone, crystallizes in P space group with two DBA18C6 (VII) and one PTA moiety in the asymmetric unit, and has a layered structure containing a water–crown ether inclusion. These waters are incorporated within the cavity of the protonated 18-crown-6 by N–H⋯O and O–H⋯O hydrogen bonds. These inclusions are aggregated via C–H⋯π interactions (aromatic interaction, T-shape), resulting in a 1D chain (Fig. 4f). The chains are further connected through the C–H⋯O, C–H⋯π and π⋯π interactions into a sheet (ESI: Tables S9 and S10†). In this 2:1 structure, both of the carboxyl groups of PTA are deprotonated. Hydrated anion layers are connected with cation crown ether layers via O–H(water)⋯O(crown), C–H(crown ether)⋯O(water), and C–H(crown ether)⋯O(PTA) hydrogen bonds (Fig. 4i). The conformation of the macrocycle and the arrangement of the pendent arm are similar to complex 5.
ΔpKa and the ionization state
Studies on protonation–deprotonation behavior in carboxylic acid–amine complexes should be helpful to distinguish if these complexes are salts or co-crystals.30 The nature (co-crystal/salt) of the complex is generally determined by the location of the H atom and the bond lengths of the two C–O bonds in the carboxylic group. The carbon–oxygen bond distances of the carboxylic group (Table 1) in exocyclic complex structures were not consistent with the deprotonated form (the C–O distances range within 1.240–1.278 Å). The evaluation can be also carried out by using FT-IR spectroscopy to observe O–H, N–H, and COOH signals and IR peak shifts due to hydrogen bonding.12b,31 When a salt is formed with amine bases, the carbonyl bands (IR bands) are shifted to lower frequencies by 30 to 40 cm−1, but in cocrystals the carbonyls are shifted due to hydrogen bonding; the magnitude of the shift is relatively small (about 10 to 20 cm−1). The IR data for complexes 1, 2 and 3 reported here indicates that each shows the presence of ∼1645 cm−1 (COO−), ∼2750 cm−1 (NH+) and of ∼1685 cm−1 (COOH) indicating partial deprotonation of the acid moiety, but complex 4 shows the presence of 1687 cm−1, and the absence of 1645 cm−1 and ∼2750 cm−1 (Fig. S1 and S2†). The Fourier difference maps revealed that the acidic protons are partially positioned in proximity to the N-atom of the aza-crown ether molecule in exocyclic complexes 1, 2 and 3. The correlation between IR data and the nature of the crown ether complexes is evident where both IR and single-crystal diffraction data exist. So the complexes 1, 2 and 3 were classified as partial proton-transfer complexes, while there is no obvious proton transfer in the co-crystal of 4. So, in figures of exocyclic complexes 1, 2, 3 and 4, we draw O–H⋯N hydrogen bonds, but all of the mono-sidearm DBA18C6s in complexes 5, 6 and 7 are totally protonated. Hence under the same crystallization condition, macrocycle bearing different structures or number of pendent arms with similar pKa, show different ionization properties. This indicate that the proton transfer depends also upon the solid state environment.| Structure no. | DC–O(short)/Å | DC–O(long)/Å | ΔDC–O/Å |
|---|---|---|---|
| 1 | 1.221 | 1.315 | 0.094 |
| 2 | 1.224 | 1.299 | 0.075 |
| 3 | 1.224 | 1.297 | 0.073 |
| 4 | 1.221 | 1.312 | 0.091 |
Exocyclic assembly behaviors by di-sidearm DBDA18C6s
Obviously, in compounds I, III and complexes 1, 2, 3 and 4, the oxygen atoms in the crown are arranged in an endo-dentate mode typical for most crowns.17b In the exocyclic complexes 1, 2, 3 and 4, similar to the compounds I and III the two nitrogen atoms extend out in a trans-fashion, deviating from the phenylate plane (C5/C6/C7/C8/C9/C10/O3/O4b) at ±0.579 Å, ±0.697 Å, ±0.659 Å, ±0.442 Å for 1, 2, 3 and 4 respectively. From the torsion angles (Table S1†), we can see that the phenylate plane (C5/C6/C7/C8/C9/C10/O3/O4b) of the crown and one CH2 unit (C11) are almost located on the same plane reference as plane R, in line with the specialty of semi-rigid macrocycle. Two phenylate planes in one macrocycle are not really in the same plane but in parallel in fact, and show less perpendicular distance through comparing the structures of compounds I, III and complexes 1, 3 respectively. The planar PTA molecule is inclined at an angle of 9.11(6)°, 15.27(7)°, 12.73(6)°, 17.75(8)° to the mean plane R for 1, 2, 3, 4. As a result, the di-sidearm macrocycles adopt the chair-shaped conformation and side arms with the axial position, similar to the structures of free macrocycle I and III. Consequently the crowns adopt a typical ‘parallelogram’ conformation17a (two phenylate planes in parallel) and nitrogen atoms in the macrocycle adopt an exo-orientation of the nitrogen lone pairs relative to the exterior by the torsions table; moreover two inversion-related methylene hydrogens turned inward toward the center of the ring. Obviously, the PTA with its hydrogen atom is involved in O–H⋯N hydrogen bonding of the head-to-head type, that results in exocyclic 1D structures like a linear ribbon. Furthermore, two methylene moieties in the macrocycle turn towards the O–C group of the PTA with their hydrogen atom being involved in C–H⋯O hydrogen bonding with the PTA. Thus synthon A (Fig. 5), assembled by O1(carboxyl)–H⋯N1(DBDA18C6) and C(DBDA18C6)–H⋯O2(carbonyl) hydrogen bonds, controls supramolecular assembly in the crystal structures of PTA and N,N′-disubstituented DBDA18C6s. In exocyclic complexes 1, 2, 3 and 4, the PTA molecule attached to the crown group with the head-to-head type, providing the exo-cyclic structures as motif A instead of motif B. There is poor information about the motif A of the aza-crown ethers and guests in the solid phase. Motif B has been illustrated15d,16 by aza-crown ethers bearing phenolic side arms with acids. | ||
| Fig. 5 The hydrogen bond synthons that appear in exocyclic complexes 1, 2, 3 and 4 ((a) O1–H⋯N1, (b) C13–H13A⋯O2, (c) C11–H11A⋯O2#, (d) C15–H15B⋯O2#, (e) C12–H12B⋯O4#, (f) C13–H13B⋯O3#, symmetry codes see ESI Tables S3–S6†). | ||
Motif B has been shown in aza-crown ethers with a flexible macrocycle and rigid pendent arms that result from steric effects,18a,22 anomeric effects32 and the hybrid method. However, in di-sidearm DBDA18C6s the anomeric effect must resulting in a side arm with an axial position to the torsion of the crown rings.33 Thus, the motif A effect could result in a side arm with an axial position to the torsions of the crown rings.33 Furthermore, the motif A in our work may be due to the orientation of the nitrogen lone pairs and the semi-rigidity of the DBDA18C6 ring as well as the flexibility of the pendent arms as mentioned above. While the conformations of the asymmetric part of the pendent arms have turned out differently, for I: trans, trans and III: trans, trans, trans; 1: trans, trans and 2: gauche, trans; for 3: gauche, trans, trans and for 4: trans (all beginning from N1 atom). It seems that the C14(macrocycle)–H⋯O5(pendent arm) intramolecular hydrogen bonding in complexes 2 and 3 can account for these disparities.
The 1D motif A ribbons are packed in a parallel fashion along the b axis in complexes 1, 2, 4 and the a axis in 3 in an ‘inclined staircase’ packing motif. It is unambiguous that the weak intermolecular C–H⋯O hydrogen bonds, which consist of two parts: C(pendent arm)–H⋯O–C(PTA) and C(macrocycle)–H⋯O(macrocycle), control the arrangement along the b axis in 1, 2, 4 and the a axis in 3 providing synthon B (Fig. 5). For 2 the distance of C13–H13B⋯O3# is 2.702 Å > 2.5 Å, instead of the strong O6–H6A⋯O2 (dH⋯O = 2.16 Å) hydrogen bonding interaction (Fig. 2). Thus, the synthon B would also benefit from pendent arms with an axial position.
Furthermore, 2D layers of crystal would pack to the 3D network. As shown in Fig. 3, different 3D packing structures could be formed together with the weak π⋯π(offset, face-to-face) or CH⋯π(point-to-face) interaction, caused by the different pendent arms. The “competition” between different functional groups in forming particular intermolecular interactions influences the 3D packing structures. The mutual arrangement of the layers of complex 3 may be attributed to a ‘staircase’ packing motif where adjacent ‘staircases’ are arranged in parallel, which caused by the π⋯π(offset face-to-face) stacking. However, in the complexes 1, 2, 4 and compound I the staircases are arranged in a herringbone-type packing due to the aromatic C–H⋯π(T-shape) interactions. Generally, as for the benzene dimers, geometry a (aromatic CH/π hydrogen bond; but this is often referred to as the point-to-face or T-shape aromatic interaction) is more favorable than geometry (b) (offset π/π stacking), though only slightly (Fig. 6). Curiously, the crystal structures of 2 and 3 reveal obvious disparities, for no other aromatic CH/π hydrogen bond or π⋯π(offset, face-to-face) interactions have been found in compound III. Thus, the structure of the pendent arms further affect the supramolecular structure based on N,N′-disubstituented DBDA18C6 bearing the pendent arms. With the length of pendent arm less than five carbon atoms, di-sidearm DBDA18C6s in the crystal lattice are apt to arrange in herringbone-type packing through the aromatic CH/π hydrogen bonding (T-shape). Meanwhile, no proof in these crystals has shown that the constitution of pendent arms in di-sidearm DBDA18C6s affects the 3D packing structure.
 | ||
| Fig. 6 Arrangement of crown ethers in the crystal lattice of compound I and complexes 1, 2 and 4 through the aromatic CH⋯π(point-to-face, T-shape) interactions between the adjacent layers (top). The weak π⋯π(offset, face-to-face) interactions between the adjacent layers in complex 3 cause a parallel packing arrangement (bottom). | ||
In addition, the interlayer C(pendent arm)–H⋯π(PTA) hydrogen bonding interactions between pendent and PTA in 2 and 4 are stronger than in 1 and 3, as the perpendicular distances of H to the ring plane are different (1, 3.08 Å; 2, 2.83 Å; 3, 3.09 Å; and 4, 2.82 Å). The reason is perhaps that the C(pendent arm)–H⋯O5(water) hydrogen bondings causes the pendent arm chain to be more curled in crystal structure 2 favoring a suitable length to form C(pendent arm)–H⋯π(PTA) hydrogen bonds as well as in solid state 4. Unfortunately, under the same conditions, we could not get similar exocyclic complexes with longer pendent arms as PTA precipitates out first (R = CH2CH2OCH2CH2CH2CH3, CH2CH2OCH2CH2OCH2CH2CH2CH3).
endo-Coordinated water salts by mono-sidearm DBA18C6s
Due to the hydrogen bonding with the water molecule in the cavity of the crown ether in salts 5, 6 and 7, the oxygen atoms and nitrogen atoms in the crown are both arranged in an endo-dentate mode, and the macrocyclic entities adopt the C-shape conformation.17b The pattern observed here shows a similar layered structure in salts 5, 6 and 7 with a sheet of protonated crown ether inclusions sandwiched between solvated PTA anion layers. Interactions between the solvated PTA anion layers and crown ethers are all through C–H⋯O hydrogen bonging in salts 5, 6 and 7, but with additional aromatic CH/π hydrogen bonding in salt 6, O–H(water)⋯O hydrogen bonding in salt 7. The structures of salts 5, 6 and 7 indicate that in these solids the supramolecular self-assembly of the ion pairs is not governed by strong and directional hydrogen-bonding interactions, and stronger and not so-directional electrostatic interactions between the cations and anions, reveal significant differences with exocyclic complexes 1, 2, 3 and 4. The observations are consistent with the fact that the “competition” between different functional groups in forming particular intermolecular interactions influences the design of supramolecular structures. It may be noted here that the each packing of the protonated crown ethers is not identical in salts 5, 6 and 7, even resulting in a similar cationic sheet. In this sheet, salt 5 shows reasonable C–H(ethyleneoxy units)⋯π and π⋯π interactions, and salt 6 shows C–H(ethyleneoxy units)⋯π without π⋯π interactions. Meanwhile, salt 7 shows aromatic CH/π hydrogen bonds and π⋯π interaction as well as C–H⋯O hydrogen bonds.The pendent arms bond to the bridge N atoms with an axial position in the structures of salts 5, 6 and 7, however the arrangement of the pendent arms is in the opposite direction between salts 5 and 6 and salt 7 (Fig. 4). The reasons for inconsistent results in salt 6 may be the N–H⋯O intramolecular hydrogen bond between the sidearm with the protonated crown ether [N1A–H1A⋯O6A = 2.873(6) Å, N1B–H1B⋯O6B = 2.894(6) Å] as well as the conformation with smaller steric hindrance in the crystal structure. As we mentioned above, with longer pendent arms (R = CH2CH2OCH2CH2CH2CH3), the crystal of PTA with di-sidearm DBDA18C6 could not be obtained, but salt 6 has been obtained. This indicates that these side arms are flexible with their position dependent upon the solid state environment; on the other hand the structure of the sidearm also affects the crystal structure.
Along with the diverse types of PTA–azacrown ether interactions, the complexes discussed herein demonstrate the different mutual arrangement of the components, which changes from the sidearm conformation in the cyclic heteroatoms and the skeleton of macrocycle to a radically different arrangement. This observation demonstrate the remarkable reliability of the N⋯O interaction between the cyclic amines and aromatic carboxylates and the promising potential for the design of multiple component crystals on the basis of such interactions. Meanwhile, the pendent arms affect the supramolecular structure of the aza-crown ether with acid: an exocyclic structure with di-sidearm DBDA18C6s but endo-coordinated water salts with mono-side arm DBA18C6s.
Conclusions
X-Ray studies for the exocyclic behavior of N,N′-disubstituented dibenzo-diaza-crown ethers, and endo-coordinated water structure of N-substituted dibenzo-aza-crown ethers with the same conditions reveal that the importance of direct strong hydrogen bonding interaction and no-direct weak hydrogen bonding in the supramolecular self-assemblies. The head-to-head arrangement of 1,4-dicarboxybenzene via O–H⋯N hydrogen bonding as a robust supramolecular motif is preserved in exocyclic supramolecular structures by the ‘out–out’ di-sidearm DBDA18C6s. However, a series of layered structures were obtained by mono-sidearm DBA18C6s with a sheet of protonated crown ether inclusions sandwiched between solvated PTA anion layers; self-assembly of the ion pairs is not governed by strong and directional hydrogen-bonding interactions or by stronger and not so-directional electrostatic interactions between the cations and anions. Moreover, the length of the side chain arm may introduce minor crystal structure disparity in di-sidearm crown ether complexes, particularly 3D packing types with a herringbone-type pattern via aromatic CH/π hydrogen bonding in complexes 1, 2, 4, but parallel packing in complex 3. The conformations of the mono-sidearm crown ether in the salts 5, 6 and 7 were influenced by the side chain chemical composition and molecular structure more than the side chain length.The ionization states of carboxylic acid–DBDA18C6 complexes are correspondingly different, and the protonation–deprotonation behavior in complexes 1, 2, 3 and co-crystal 4 are clear from the single-crystal X-ray diffraction analyses and FT-IR data. Furthermore, the assembly behavior of these lariat ether may also be affected by the guests, and these are also currently under investigation.
Experimental
Crystallization
The azacrown ethers (I, II, III, IV, V, VI, VII) were prepared according to the literature method.16a,16b Single crystals were grown from CH2Cl2–hexane mixtures for compounds I, II, III, IV, while only crystals of compounds I and III are available. Azacrown ethers (I, II, III, IV, V, VI) and PTA were at a 1:1 (acid:amine) molar ratio in a water–acetone medium, but VII and PTA was at 2:1. The resultant mixture was subjected to reflux for two hours to ensure the homogeneous mixing of the two components. A colorless precipitate was obtained after complete removal of solvent by rotavapor. Single crystals were grown from water–acetone mixtures (25 mg of complexes in 4 mL solvents in 10 mL beaker) by slow evaporation at room temperature. Typically X-ray quality crystals appeared after a few weeks.
IR spectroscopy
Transmission infrared spectra of the solids were obtained using a Fourier-transform infrared spectrometer (Nicolet NEXUS670). 16 scans were collected at 4 cm−1 resolution for each sample. The spectra were measured over the range of 4000–400 cm−1.X-Ray crystallography
X-Ray data of the crystals were collected on a Bruker D8 Quest CMOS single-crystal diffractometer with graphite filtered Mo Kα (λ = 0.71073 Å) radiation. Data collections for crystals of I, 1, 2, 3, 4, 5, 6 and 7 were carried out at 173(2) K and III at 298(2) K. The structures were solved by direct methods using the SHELXS-9734 program and refined by fullmatrix least-squares on F2 using SHELXL97.34 H atoms attached to O and N parents were found in the Fourier maps and refined with distance restraints, and referring to complexes 1, 2 and 3, these H atoms were refined with a split atom model for the discussion of the behavior on proton transfer. Diagrams and publication material were generated using WinGX,35 ORTEP,36 and PLATON.37 Crystallographic data and structural correction parameters are listed in Table 2.
| Compound reference | I | III | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|---|---|
| CCDC number | 1058279 | 1058280 | 1048852 | 1048853 | 1048854 | 1048855 | 1060647 | 1060648 | 1060649 |
| Chemical formula | C28H42N2O4 | C28H42N2O6 | C28H42N2O4·C8H6O4 | C26H38N2O6·C8H6O4·0.2(H2O) | C28H42N2O6·C8H6O4 | C26H34N2O4·C8H6O4 | 3(C24H34NO5)·3(C8H5O4)·10(H2O) | C26H37NO6· C8H5O4·4(H2O) | 2(C25H36NO5)·C8H4O4·13(H2O) |
| Formula mass | 470.64 | 502.64 | 636.76 | 644.31 | 668.76 | 604.68 | 1925.09 | 697.76 | 1259.42 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Monoclinic | Triclinic | Monoclinic | Triclinic | Orthorhombic | Triclinic |
| a/Å | 16.1140(18) | 4.8922(15) | 31.850(3) | 31.427(4) | 5.7584(5) | 31.022(6) | 15.5676(9) | 15.4985(8) | 12.9024(13) |
| b/Å | 4.7953(5) | 10.102(3) | 5.7811(5) | 5.8115(8) | 10.5829(9) | 5.8603(15) | 16.2241(8) | 18.7452(11) | 12.9804(16) |
| c/Å | 17.101(2) | 14.454(4) | 20.098(2) | 19.863(2) | 14.5837(13) | 19.725(4) | 21.6195(13) | 25.1312(15) | 31.537(4) |
| α/° | 90.00 | 81.084(5) | 90.00 | 90.00 | 103.242(3) | 90.00 | 85.854(2) | 90.00 | 82.392(4) |
| β/° | 97.747(4) | 87.770(5) | 112.557(3) | 114.729(4) | 95.682(3) | 118.646(6) | 82.127(2) | 90.00 | 82.392(4) |
| γ/° | 90.00 | 84.859(5) | 90.00 | 90.00 | 100.938(3) | 90.00 | 66.1610(10) | 90.00 | 82.392(4) |
| Unit cell volume/Å3 | 1309.3(3) | 702.7(4) | 3417.4(6) | 3295.2(7) | 839.90(13) | 3146.9(12) | 4946.6(5) | 7301.2(7) | 4861.4(10) |
| Temperature/K | 173(2) | 298(2) | 173(2) | 173(2) | 173(2) | 173(2) | 173(2) | 173(2) | 173(2) |
| Space group | P2(1)/n | P |
C2/c | C2/c | P |
C2/c | P |
P2(1)2(1)2(1) | P |
| No. of formula units per unit cell, Z | 2 | 1 | 4 | 4 | 1 | 4 | 2 | 8 | 3 |
| No. of reflections measured | 6253 | 4942 | 9043 | 7854 | 6796 | 10964 |
38605 |
33278 |
39452 |
| No. of independent reflections | 2273 | 2446 | 3090 | 2938 | 2988 | 2841 | 17369 |
7735 | 16713 |
| Rint | 0.0749 | 0.0448 | 0.0468 | 0.0750 | 0.0514 | 0.0845 | 0.0974 | 0.0920 | 0.1183 |
| Final R1 values (I > 2σ(I)) | 0.0563 | 0.0552 | 0.0475 | 0.0519 | 0.0509 | 0.0513 | 0.0622 | 0.0569 | 0.0872 |
| Final wR(F2) values (I > 2σ(I)) | 0.1144 | 0.1683 | 0.1129 | 0.0888 | 0.1029 | 0.1244 | 0.0920 | 0.1307 | 0.1376 |
| Final R1 values (all data) | 0.1226 | 0.0687 | 0.0932 | 0.1305 | 0.1091 | 0.1144 | 0.1714 | 0.1388 | 0.2170 |
| Final wR(F2) values (all data) | 0.1406 | 0.1827 | 0.1364 | 0.1113 | 0.1221 | 0.1816 | 0.1187 | 0.1702 | 0.1778 |
Acknowledgements
We are grateful to the National Natural Science Foundation of China (20772092) and the Hubei Province Natural Science Fund for Distinguished Young Scholars (2007ABB021) for financial support.Notes and references
- G. R. Desiraju, Nature, 2001, 412, 397–400 CrossRef CAS PubMed.
- J. C. MacDonald and G. M. Whitesides, Chem. Rev., 1994, 94, 2383–2420 CrossRef CAS.
- A. Ballabh, D. R. Trivedi and P. Dastidar, Cryst. Growth Des., 2005, 5, 1545–1553 CAS.
- (a) G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS PubMed; (b) P. Ganguly and G. R. Desiraju, CrystEngComm, 2010, 12, 817–833 RSC; (c) A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- (a) L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS; (b) K. Sada, T. Tani and S. Shinkai, Synlett, 2006, 2364–2374 CrossRef CAS; (c) Y. Li, W. Yang, R. Guo, Y. Chen and S. Gong, CrystEngComm, 2012, 14, 1455–1462 RSC.
- (a) P. Sahoo and P. Dastidar, Cryst. Growth Des., 2012, 12, 5917–5924 CrossRef CAS; (b) A. Lemmerer, CrystEngComm, 2011, 13, 2849 RSC; (c) I. Hisaki, T. Sasaki, N. Tohnai and M. Miyata, Chem.–Eur. J., 2012, 18, 10066–10073 CrossRef CAS PubMed.
- A. Tanaka, K. Inoue, I. Hisaki, N. Tohnai, M. Miyata and A. Matsumoto, Angew. Chem., 2006, 118, 4248–4251 CrossRef PubMed.
- (a) K. Megumi, F. Nadiah Binti Mohd Arif, S. Matsumoto and M. Akazome, Cryst. Growth Des., 2012, 12, 5680–5685 CrossRef CAS; (b) A. Ichikawa, H. Ono, T. Echigo and Y. Mikata, CrystEngComm, 2011, 13, 4536–4548 RSC.
- (a) A. D. Bond, Cryst. Growth Des., 2005, 5, 755–771 CrossRef CAS; (b) L. Chen, H. Y. Zhang and Y. Liu, J. Org. Chem., 2012, 77, 9766–9773 CrossRef CAS PubMed; (c) Y. Liu and Y. Chen, Acc. Chem. Res., 2006, 39, 681–691 CrossRef CAS PubMed.
- (a) T. Yuge, T. Sakai, N. Kai, I. Hisaki, M. Miyata and N. Tohnai, Chem.–Eur. J., 2008, 14, 2984–2993 CrossRef CAS PubMed; (b) T. Sasaki, Y. Ida, A. Tanaka, I. Hisaki, N. Tohnai and M. Miyata, CrystEngComm, 2013, 15, 8237 RSC.
- (a) X. Li, S.-L. Gong, W.-P. Yang, Y.-Y. Chen and X.-G. Meng, Tetrahedron, 2008, 64, 6230–6237 CrossRef CAS PubMed; (b) Y. Li, W. Yang, Y. Chen and S. Gong, CrystEngComm, 2011, 13, 259–268 RSC.
- (a) S. Y. Han and H. Bin Oh, Chem. Phys. Lett., 2006, 432, 269–274 CrossRef CAS PubMed; (b) S. L. Childs, G. P. Stahly and A. Park, Mol. Pharmaceutics, 2007, 4, 323–338 CrossRef CAS PubMed; (c) S. Bhattacharya and B. K. Saha, Cryst. Growth Des., 2011, 11, 2194–2204 CrossRef CAS.
- (a) R. Kaur, S. S. R. R. Perumal, A. J. Bhattacharyya, S. Yashonath and T. N. Guru Row, Cryst. Growth Des., 2014, 14, 423–426 CrossRef CAS; (b) S. Kohmoto, S. Sekizawa, S. Hisamatsu, H. Masu, M. Takahashi and K. Kishikawa, Cryst. Growth Des., 2014, 14, 2209–2217 CrossRef CAS.
- (a) N. Stanley, V. Sethuraman, P. T. Muthiah, P. Luger and M. Weber, Cryst. Growth Des., 2002, 2, 631–635 CrossRef CAS; (b) X.-C. Su, H.-K. Lin, S.-R. Zhu, L.-H. Weng, X.-B. Leng and Y.-T. Chen, Supramol. Chem., 2002, 14, 41–45 CrossRef CAS PubMed; (c) S. B. Raj, N. Stanley, P. T. Muthiah, G. Bocelli, R. Ollá and A. Cantoni, Cryst. Growth Des., 2003, 3, 567–571 CrossRef CAS; (d) X.-L. Zhang and X.-M. Chen, Cryst. Growth Des., 2005, 5, 617–622 CrossRef CAS.
- (a) B. Sarma and B. Saikia, CrystEngComm, 2014, 16, 4753 RSC; (b) J. B. Nanubolu, B. Sridhar, K. Ravikumar, K. D. Sawant, T. A. Naik, L. N. Patkar, S. Cherukuvada and B. Sreedhar, CrystEngComm, 2013, 15, 4448 RSC; (c) R. Montis and M. B. Hursthouse, CrystEngComm, 2012, 14, 7466 RSC; (d) S. Ghosh, P. P. Bag and C. M. Reddy, Cryst. Growth Des., 2011, 11, 3489–3503 CrossRef CAS.
- (a) V. J. Gatto and G. W. Gokel, J. Am. Chem. Soc., 1984, 106, 8240–8244 CrossRef CAS; (b) N. G. Lukyanenko, S. S. Basok and L. K. Filonova, J. Chem. Soc., Perkin Trans. 1, 1988, 3141, 10.1039/p19880003141; (c) V. Rüdiger, H.-J. Schneider, V. P. Solov’ev, V. P. Kazachenko and O. A. Raevsky, Eur. J. Org. Chem., 1999, 1847–1856 CrossRef; (d) P. Wei, X. Yan and F. Huang, Chem. Soc. Rev., 2015, 44, 815–832 RSC; (e) S. Dong, B. Zheng, F. Wang and F. Huang, Acc. Chem. Res., 2014, 47, 1982–1994 CrossRef CAS PubMed; (f) Y. Gao, R.-L. Zhong, H.-L. Xu, S.-L. Sun and Z.-M. Su, RSC Adv., 2015, 5, 30107–30119 RSC; (g) D. Liu, T. Pang, K. Ma, W. Jiang and X. Bao, RSC Adv., 2014, 4, 2563–2567 RSC.
- (a) G. W. Gokel, L. J. Barbour, R. Ferdani and J. Hu, Acc. Chem. Res., 2002, 35, 878–886 CrossRef CAS PubMed; (b) G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 CrossRef CAS PubMed; (c) S. J. Pond, O. Tsutsumi, M. Rumi, O. Kwon, E. Zojer, J. L. Bredas, S. R. Marder and J. W. Perry, J. Am. Chem. Soc., 2004, 126, 9291–9306 CrossRef CAS PubMed; (d) E. Kleinpeter and A. Holzberger, Tetrahedron, 2006, 62, 10237–10247 CrossRef CAS PubMed; (e) H. Tomiyasu, J.-L. Zhao, X.-L. Ni, X. Zeng, M. R. J. Elsegood, B. Jones, C. Redshaw, S. J. Teat and T. Yamato, RSC Adv., 2015, 5, 14747–14755 RSC; (f) H.-L. Lee, N. Dhenadhayalan and K.-C. Lin, RSC Adv., 2015, 5, 4926–4933 RSC; (g) N. Gutowska, P. Seliger, G. Andrijewski, M. Siwy, M. Małecka and J. Kusz, RSC Adv., 2015, 5, 38435–38442 RSC; (h) X. Bao, D. Liu, Y. Jin, X. Liu and W. Jiang, RSC Adv., 2013, 3, 6783 RSC; (i) M. Sarma, T. Chatterjee and S. K. Das, RSC Adv., 2012, 2, 3920 RSC.
- (a) M. S. Fonari, E. V. Ganin, S. S. Basok, K. A. Lyssenko, M. J. Zaworotko and V. C. Kravtsov, Cryst. Growth Des., 2010, 10, 5210–5220 CrossRef CAS; (b) J. M. Cole, P. G. Waddell and D. Jayatilaka, Cryst. Growth Des., 2012, 12, 2277–2287 CrossRef CAS.
- S. Park, S. Y. Lee, K. M. Park and S. S. Lee, Acc. Chem. Res., 2012, 45, 391–403 CrossRef CAS PubMed.
- H. E. Simmons and C. H. Park, J. Am. Chem. Soc., 1968, 90, 2428–2429 CrossRef CAS.
- D. J. Wolstenholme, P. Sirsch, L. Onut, J. Flogeras, A. Decken and G. S. McGrady, Dalton Trans., 2011, 40, 8301 RSC.
- M. S. Fonari, E. V. Ganin, Y. M. Chumakov, M. M. Botoshansky, K. Suwinska, S. S. Basok and Y. A. Simonov, New J. Chem., 2009, 33, 1646 RSC.
- J.-C. Aguilar, S. Bernès, P. Gómez-Tagle, R. A. Bartsch and J. de Gyves, J. Chem. Crystallogr., 2006, 36, 473–479 CrossRef CAS.
- J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed.
- M. B. Duriska, S. M. Neville and S. R. Batten, Chem. Commun., 2009, 5579–5581 RSC.
- B. R. Bhogala, P. Vishweshwar and A. Nangia, Cryst. Growth Des., 2005, 5, 1271–1281 CAS.
- (a) E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CAS; (b) G. R. Desiraju, Acc. Chem. Res., 1991, 24, 290–296 CrossRef CAS.
- (a) S. Tsuzuki, Annu. Rep. Prog. Chem., Sect. C, 2012, 108, 69 RSC; (b) M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873 RSC.
- (a) C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS; (b) C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC; (c) R. R. Choudhury and R. Chitra, CrystEngComm, 2010, 12, 2113 RSC.
- (a) V. R. Hathwar, R. Pal and T. N. Guru Row, Cryst. Growth Des., 2010, 10, 3306–3310 CrossRef CAS; (b) T. R. Shattock, K. K. Arora, P. Vishweshwar and M. J. Zaworotko, Cryst. Growth Des., 2008, 8, 4533–4545 CrossRef CAS; (c) S. Mohamed, D. A. Tocher, M. Vickers, P. G. Karamertzanis and S. L. Price, Cryst. Growth Des., 2009, 9, 2881–2889 CrossRef CAS; (d) B. Sarma, N. K. Nath, B. R. Bhogala and A. Nangia, Cryst. Growth Des., 2009, 9, 1546–1557 CrossRef CAS.
- C. B. Aakeroy, J. Desper and M. E. Fasulo, CrystEngComm, 2006, 8, 586–588 RSC.
- G. R. J. Thatcher, The Anomeric Effect and Associated Stereoelectronic Effects, American Chemical Society, Washington, 1993 Search PubMed.
- M. W. Hosseini, A. J. Blacker and J. M. Lehn, J. Am. Chem. Soc., 1990, 112, 3896–3904 CrossRef CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (b) G. M. Sheldrick, SHELXL-97, A Program for Crystal Structure Solution, University of Göttingen, Germany, 1997 Search PubMed; (c) G. M. Sheldrick, SHELXL-97, A Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1 and S2: torsion angles of macrocyclic rings and the pendent arms; Tables S3–S9: hydrogen bonds of supramolecular complexes 1–7; Table S10a: CH⋯π hydrogen bonds, Table S10b: π⋯π interactions. Table S11: TGA and DSC. Table S12 (Fig. S1 and S2): FT-IR data. Crystallographic information file (cif). CCDC 1048852–1048855, 1058279, 1058280, 1060647–1060649. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra09521b |
| This journal is © The Royal Society of Chemistry 2015 |