
Ultrasonic-assisted biodiesel production from waste cooking oil over novel sulfonic functionalized carbon spheres derived from cyclodextrin via one-step: a way to produce biodiesel at short reaction time†
Abstract
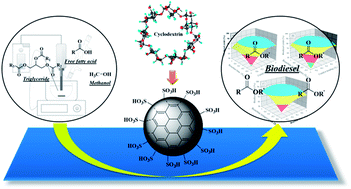
In this study, a novel sulfonated carbon catalyst was synthesized via the one-step hydrothermal carbonization of cyclodextrin, hydroxyethylsulfonic acid and citric acid. Ultrasonic-assisted biodiesel production from waste cooking oil in the presence of the catalyst was investigated. The novel catalyst was characterized by BET, XRD, PSD, SEM-EDS, TGA, FT-IR, XPS and TPD. The catalyst exhibited a high acidity of up to 1.87 mmol g−1. 2k factorial and Box–Behnken designs were applied to find the optimum conditions to obtain a maximum fatty acid methyl ester (FAME) yield. The results of the optimization imply that a catalyst loading of 11.5 wt%, a reaction time of 8.8 min and a reaction temperature of 117 °C provide a maximum FAME yield of up to 90.8% in ultrasonic-assisted biodiesel production. The reusability of the catalyst was studied for 4 cycles under the optimum conditions and the results showed that the regenerated catalyst can be reused without any serious reduction of the FAME yield. Kinetic studies showed that the reaction followed first order reaction kinetics with an activation energy of 11.64 kJ mol−1.
 Please wait while we load your content...
Please wait while we load your content...

