
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coating of bioactive glass particles with mussel-inspired polydopamine as a strategy to improve the thermal stability of poly(L-lactide)/bioactive glass composites†
Aitor
Larrañaga‡
*,
Daniel
Ramos‡
,
Hegoi
Amestoy
,
Ester
Zuza
and
Jose-Ramon
Sarasua
University of the Basque Country (UPV/EHU), Department of Mining-Metallurgy Engineering and Materials Science & POLYMAT, School of Engineering, Alameda de Urquijo s/n, 480130 Bilbao, Spain. E-mail: aitor.larranagae@ehu.eus; Fax: +34 94 601 3930; Tel: +34 946 017 138
First published on 31st July 2015
Abstract
Fabrication of bioactive glass (BG) filled polyester composites by traditional thermoplastic processing techniques is limited because a thermal degradation reaction occurs between the Si–O− groups present on the surface of the BG and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups present in the backbone of the polymer at high temperatures. To overcome this problem, this study looked at the effect of mussel-inspired polydopamine (PDA) coated on the surface of BG particles. Thermogravimetric analysis demonstrated the improved thermal stability of poly(L-lactide) (PLLA) composites filled with PDA-coated BG particles compared to PLLA composites filled with uncoated BG particles. Moreover, these composites were successfully manufactured by hot-pressing, showing enhanced mechanical properties in comparison to non-coated BG filled PLLA composites. Dynamic mechanical analysis indicated a good interfacial interaction between PDA-coated BG particles and the PLLA matrix, suggesting that an immobilized layer of polymer chains was formed around the BG particles. Finally, the bioactivity of PLLA samples filled with 15 vol% of PDA-coated BG particles was confirmed via the deposition of an apatite layer on the surface of the material. In view of the results obtained it can be concluded that coating BG particles with PDA is a promising strategy as it can create composite materials with improved thermal stability and bioactivity.
O groups present in the backbone of the polymer at high temperatures. To overcome this problem, this study looked at the effect of mussel-inspired polydopamine (PDA) coated on the surface of BG particles. Thermogravimetric analysis demonstrated the improved thermal stability of poly(L-lactide) (PLLA) composites filled with PDA-coated BG particles compared to PLLA composites filled with uncoated BG particles. Moreover, these composites were successfully manufactured by hot-pressing, showing enhanced mechanical properties in comparison to non-coated BG filled PLLA composites. Dynamic mechanical analysis indicated a good interfacial interaction between PDA-coated BG particles and the PLLA matrix, suggesting that an immobilized layer of polymer chains was formed around the BG particles. Finally, the bioactivity of PLLA samples filled with 15 vol% of PDA-coated BG particles was confirmed via the deposition of an apatite layer on the surface of the material. In view of the results obtained it can be concluded that coating BG particles with PDA is a promising strategy as it can create composite materials with improved thermal stability and bioactivity.
1. Introduction
Combination of bioactive glass (BG) particles or fibers with resorbable synthetic polymers has emerged as a promising strategy for the preparation of materials whose applications range from structural implants to tissue engineering scaffolds, and in particular for bone related biomedical applications.1 On the one hand, synthesis conditions can be finely controlled to prepare resorbable synthetic polymers (e.g., poly(L-lactide) (PLLA), poly(ε-caprolactone) (PCL) and its copolymers (PLCL)) that have tunable mechanical properties2 and degradation rates.3,4 On the other hand, incorporation of BG imparts osteoproductivity and osteoconductivity to the bioinert polymer.5 Accordingly, the ionic dissolution products from BG (particularly silica and calcium ions) activate several families of genes that control the osteoblast cycle, mitosis and differentiation, giving rise to rapid bone regeneration.6,7 Moreover, in contact with body fluids, a hydroxycarbonate apatite layer is developed on the surface of BG, this forms a strong bond with the native bone and avoids any relative motion (micromotion) of the implanted scaffold. Outside of bone related biomedical applications, recent studies have also highlighted the advantages of BG in soft tissue regeneration and wound healing.8,9Since the aforementioned synthetic (co)polymers are thermoplastic, they are typically manufactured by injection molding, blow molding, thermoforming or extrusion.10 However, previous studies have revealed that a degradation reaction between the Si–O− groups present on the surface of BG and the CO groups present in the polymer backbone occurs at high temperatures,11–13 the result is a dramatic reduction in the molecular weight of the polymer matrix. This is why several studies have reported detrimental effects on the mechanical properties of the resulting composites after the incorporation of BG, when processed by traditional thermoplastic techniques.12–15
In a previous work of our group16 a surface modification of BG particles by plasma polymerization of acrylic acid was proposed as a strategy for the improvement of the thermal stability of BG filled composite systems. Even if obtained results were promising, the homogeneity of the treatment was not optimal since it did not assure the modification of the whole surface of all particles. In the present work, coating BG particles with mussel-inspired polydopamine (PDA) as a strategy to maintain the thermal stability of PLLA/BG composites is proposed. PDA is a dopamine derived synthetic eumelanin polymer that contains both cathecol and amine functionalities in its backbone.17 In the case of previously proposed plasma polymerization strategy, plasma reactors with fluidized beds or circulating streams are needed to achieve and homogeneous coating. In contrast, in the strategy proposed in the present work, a thin and homogeneous PDA film can be deposited on the surface of the material spontaneously via simple immersion of substrates in a dilute aqueous solution of dopamine.18 Thus, many different substrates have been coated with PDA in recent years.19–24 However, to the best of the author's knowledge, PDA has not so far been employed on BG. In this study BG particles were first coated with PDA (mBG) and fully characterized by Fourier transform infrared spectroscopy (FTIR) and X-ray photoelectron spectroscopy (XPS). The thermal degradation behavior of PLLA/mBG was then studied by means of thermogravimetric analysis (TGA). After that, hot-pressed sheets were manufactured and their mechanical properties were analyzed via tensile test and dynamic mechanical analysis (DMA). Finally, these samples were submerged in simulated body fluid (SBF) and the formation of a hydroxyapatite layer was monitored by scanning electron microscopy (SEM) and X-ray diffractometry (XRD).
2. Materials and methods
2.1. Materials
PLLA with a weight-average molecular weight (Mw) of 160![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and polydispersity index of 1.7 was supplied by Biomer (Germany). 45S5 Bioglass® particles were kindly supplied by Novabone® (US), their composition being (in wt%) 45.0% SiO2, 24.4% Na2O, 24.5% CaO and 6.0% P2O5. To measure their size distribution, a dispersion of these particles in ethanol was prepared, and after sonication for 15 min a few drops were placed on a glass slide. Finally, the sample was examined under a microscope and the size distribution was determined using ImageJ software. The particle size was <60 μm, with a mean particle size of 9 μm and density of 2.75 g cm−3. Dopamine hydrochloride, tris(hydroxymethyl)aminomethane (Trizma® base), NaCl, NaHCO3, KCl, K2HPO4·3H2O, MgCl2·6H2O, CaCl2, Na2SO4 and hydrochloric acid (HCl, 37%) were purchased from Sigma-Aldrich (Spain). Chloroform was purchased from Panreac (Spain).
000 g mol−1 and polydispersity index of 1.7 was supplied by Biomer (Germany). 45S5 Bioglass® particles were kindly supplied by Novabone® (US), their composition being (in wt%) 45.0% SiO2, 24.4% Na2O, 24.5% CaO and 6.0% P2O5. To measure their size distribution, a dispersion of these particles in ethanol was prepared, and after sonication for 15 min a few drops were placed on a glass slide. Finally, the sample was examined under a microscope and the size distribution was determined using ImageJ software. The particle size was <60 μm, with a mean particle size of 9 μm and density of 2.75 g cm−3. Dopamine hydrochloride, tris(hydroxymethyl)aminomethane (Trizma® base), NaCl, NaHCO3, KCl, K2HPO4·3H2O, MgCl2·6H2O, CaCl2, Na2SO4 and hydrochloric acid (HCl, 37%) were purchased from Sigma-Aldrich (Spain). Chloroform was purchased from Panreac (Spain).
2.2. Coating of BG particles with PDA
In a typical experiment, 2 g of dopamine were dissolved in 500 mL of Tris–HCl solution (10 mM, pH = 8.5). The solution gradually turned gray due to the oxidation of the dopamine. Subsequently, 4 g of BG particles were added and the dispersion was stirred for 24 h at room temperature. Then, the surface coated BG particles, named as mBG, were collected by filtration and rinsed with distilled water until the washing liquid became colorless. Finally, the mBG particles were dried at 50 °C under reduced pressure during 48 h. It is worth noting that mBG particles showed better stability in distilled water than BG particles (Fig. S1†). The hydrophilic –OH and –NH2 groups present in PDA may facilitate the dispersion of mBG particles in aqueous solutions.2.3. Preparation of PLLA/mBG films and sheets
PLLA films containing 5, 10 and 15 vol% of BG or mBG particles were prepared by solvent casting. Predetermined amounts of BG or mBG particles were added to a PLLA solution in chloroform (4 w/v%) and vigorously stirred for 1 h. Finally, after chloroform evaporation the resulting films were dried at 50 °C under reduced pressure for 48 h in a vacuum oven.These films were cut into small pieces and processed by hot-pressing in a Collin's P200E hydraulic press as follows: once the hydraulic press reached 200 °C, the polymer was put within the press between two metal plates (mold) and kept at this temperature for 5 minutes. Then, 200 bar were applied and, after 1 minute, the metal plates were cooled down with a water refrigeration system. In this way PLLA sheets of ∼1 mm thickness containing 5, 10 or 15 vol% of BG or mBG particles were obtained.
2.4. Characterization
The Ca–P layer developed on the surface of samples was characterized using scanning electron microscopy (SEM) (HITACHI S-3400). Samples were coated with a 150 Å layer of gold in a JEL Ion Sputter JFC-1100 at 1200 V and 5 mA.
The formation and crystallization of hydroxyapatite was confirmed by means of X-ray difractometry (XRD). The X-ray powder diffraction patterns were collected by a PHILIPS X'PERT PRO automatic diffractometer operating at 40 kV and 40 mA, in theta–theta configuration, secondary monochromator with Cu-Kα radiation (λ = 1.5418 Å) and a PIXcel solid state detector. Data were collected from 10 to 50° 2θ (step size = 0.026 and time per step = 0.8 s per channel) at room temperature. A fixed divergence and anti-scattering slit to produce a constant volume of sample illumination were used.
Additionally, mBG particles were directly submerged in SBF at 37 °C. After 1, 3 and 7 days, they were collected, rinsed with distilled water and dried under reduced pressure prior to FTIR analysis.
3. Results and discussion
3.1. Coating of BG particles with PDA
The color of BG particles turned from white to gray after the coating process, indicating the formation of a layer of PDA on the surface of BG particles (Fig. 1). | ||
| Fig. 1 Picture of BG particles before (a) and after (b) PDA coating. | ||
The successful coating of PDA on the surface of BG particles was further confirmed by XPS (Fig. 2 and Table 1) and FTIR (Fig. S2†). The XPS survey spectra of BG particles (Fig. 2b) indicate the presence of O 1s (531.6 eV), C 1s (285.0 eV), Ca 2p (346.6 eV), Na 1s (1071.6 eV) and Si 2p (102.6 eV) on the surface of the particles. After PDA coating, the XPS survey spectra of mBG particles showed an additional peak at 399.6 eV, which corresponds to N 1s. The presence of nitrogen is ascribed to the amine functionalities in the PDA backbone and confirms the coating of PDA on the surface of mBG particles. The N/C signal ratio was 0.089, that is very close to the value reported for polydopamine (N/C = 0.086).24 In FTIR (Fig. S2†) the main characteristics of the spectrum of BG particles are attributed to the amorphous silica glass. The two bands at 1050 and 950 cm−1 are assigned to Si–O–Si stretching vibration and the band at 450 cm−1 is assigned to Si–O–Si bending vibration.27 In contrast, the main bands of mBG spectra appeared at 1595 and 1505 cm−1, this can be assigned to νring(CC) and νring(CN) stretching modes, respectively, indicating that aromatic amines were present in the coated PDA film. Moreover, a broad band was observed between 3700–2500 cm−1, this can be assigned to the ν(N–H) and ν(O–H) stretching modes.28
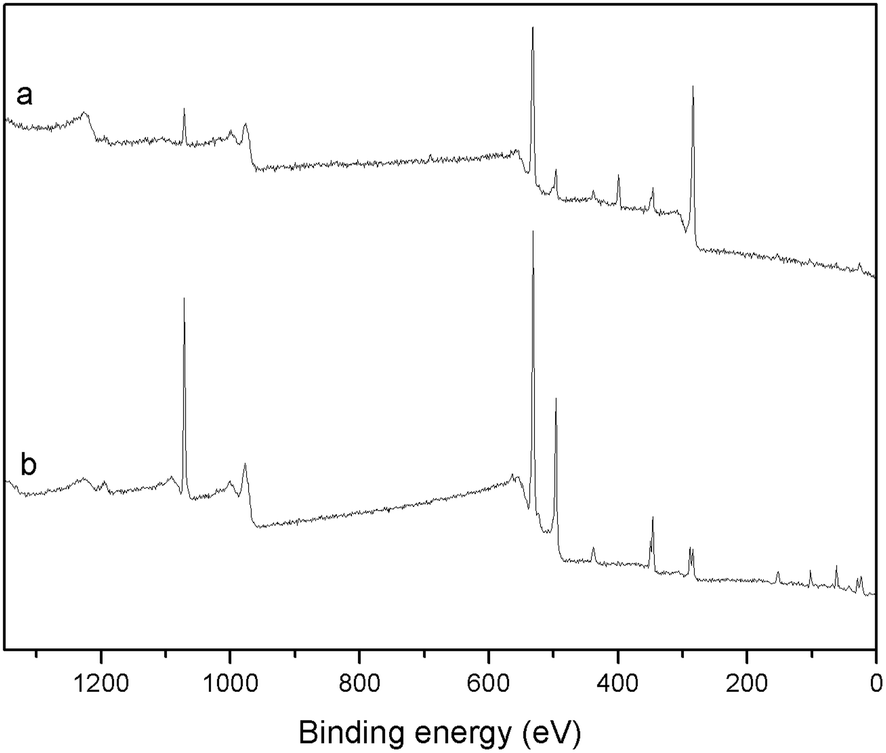 | ||
| Fig. 2 XPS survey spectra of mBG (a) and BG (b) particles. | ||
| Element | Atomic% | |
|---|---|---|
| BG particles | mBG particles | |
| O 1s | 48.2 | 22.6 |
| C 1s | 24.4 | 64.8 |
| N 1s | — | 5.8 |
| Ca 2p | 6.1 | 2.2 |
| Na 1s | 12.8 | 1.7 |
| Si 2p | 8.4 | 2.9 |
Finally, the thermal stability of BG and mBG particles up to 700 °C was analyzed by TGA (Fig. S3†). In this temperature range, BG particles did not suffer any significant weight losses. In contrast, mBG lost 13% of its initial weight at the end of the process. This weight loss can only be attributed to the presence of a PDA coating on the surface of mBG particles.
3.2. Thermal stability of PLLA/BG and PLLA/mBG composites
Fig. 3 shows the thermogravimetry (TG) and differential thermogravimetry (DTG) profiles of PLLA composites prepared by solvent casting. From this figure, peak (Tpeak) and onset (Tonset, calculated from the inflection point of the thermogravimetry profile) degradation temperatures can be determined, these are summarized in Table 2. A first weight loss (∼10% weight loss) was observed in all cases at around 100 °C that can be associated to some water present in the samples. For pristine PLLA, Tonset and Tpeak were 303.5 ± 11.5 and 338.5 ± 8.5 °C, respectively. These values decreased constantly as the content of BG particles increased. For example, the Tonset and Tpeak values for the sample containing 5 vol% of BG particles were 202.5 ± 11.5 and 277.5 ± 2.5 °C respectively. In the case of the sample filled with 15 vol% of BG particles these temperatures were 190.0 ± 1.0 and 248.0 ± 1.5 °C respectively. Moreover, the peak from the DTG profile became broader as the content of BG particles increased, suggesting a more complex degradation mechanism in those samples filled with BG particles. As previously reported,11–13 a degradation reaction between the Si–O− groups present on the surface of the particles and the CO groups in the polymer backbone occurs at high temperatures. This degradation reaction results in the random scission of PLLA polymer chains; hence, both Tonset and Tpeak dramatically decrease in the presence of BG particles. In contrast, coating PDA on the surface of mBG particles improved the thermal stability of PLLA composites. In this sense, PLLA samples containing 15 vol% of mBG particles had Tonset and Tpeak values of 282.5 ± 1.1 and 309.0 ± 1.0 °C, respectively, which are much closer to the values calculated for pristine PLLA. In addition, the peak in the DTG profile did not obviously become broader as the content of mBG particles in the composite increased, suggesting that the degradation reaction between the Si–O− groups present on the surface of the particles and the CO groups in the polymer backbone was impeded.
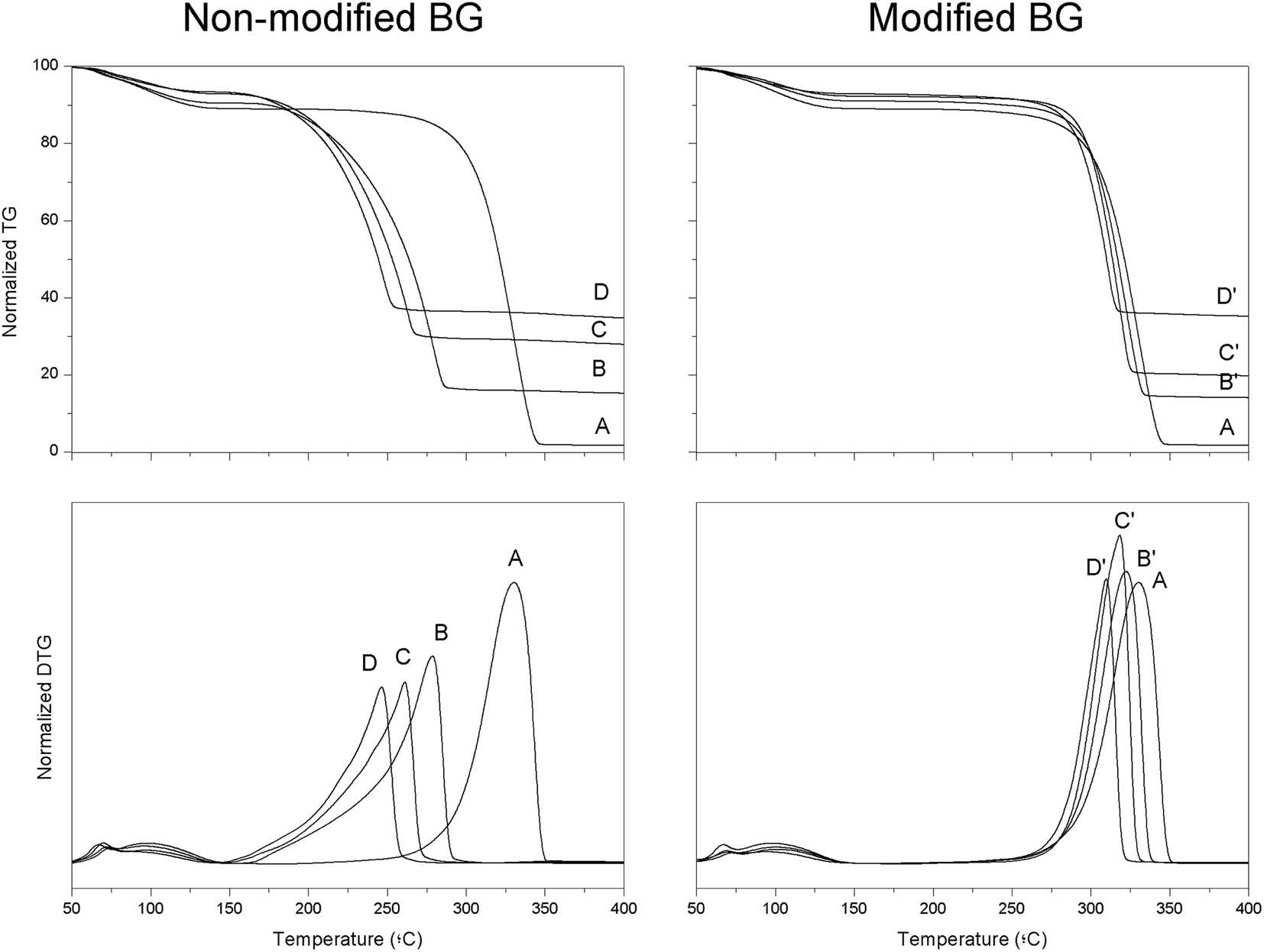 | ||
| Fig. 3 TG and DTG profiles of PLLA composites containing 5, 10 or 15 vol% of BG or mBG particles. | ||
| Sample | Vol% BG | T peak (°C) | T onset (°C) | Deg. ratea (% per min) | Final weightb (%) |
|---|---|---|---|---|---|
| a Calculated from the slope of weight loss in the isothermal experiments. b Final weight of the samples after 20 min at 210 °C. | |||||
| PLLA | 0 | 338.5 ± 8.5 | 303.5 ± 11.5 | 0.03 ± 0.02 | 89.9 ± 0.8 |
| PLLA 5BG | 7.2 ± 1.5 | 277.5 ± 2.5 | 202.5 ± 1.0 | 1.2 ± 0.1 | 67.3 ± 1.8 |
| PLLA 5mBG | 6.3 ± 0.9 | 322.5 ± 1.0 | 293.5 ± 1.0 | 0.09 ± 0.03 | 89.8 ± 0.1 |
| PLLA 10BG | 16.9 ± 2.0 | 258.5 ± 3.5 | 194.5 ± 1.5 | 1.9 ± 0.1 | 51.1 ± 0.8 |
| PLLA 10mBG | 10.2 ± 1.4 | 318.5 ± 0.5 | 291.5 ± 1.0 | 0.04 ± 0.001 | 92.1 ± 0.1 |
| PLLA 15BG | 20.4 ± 1.1 | 248.0 ± 1.5 | 190.0 ± 1.0 | 2.1 ± 0.1 | 51.3 ± 1.9 |
| PLLA 15mBG | 20.1 ± 1.8 | 309.0 ± 1.0 | 282.5 ± 1.1 | 0.03 ± 0.003 | 93.3 ± 0.1 |
Results from isothermal treatments (Fig. S4† and Table 2) also demonstrated the improved thermal stability of samples containing PDA-coated mBG particles with respect to their non-coated BG filled counterparts. For example, PLLA samples containing 15 vol% of BG particles (PLLA 15BG), lost almost 50% of their initial mass after being subjected to the isothermal treatment at 210 °C for 20 min, showing a degradation rate of 2.1 ± 0.1% per min. In contrast, the samples containing 15 vol% of mBG particles (PLLA 15mBG), only lost 6.7% of their initial mass after the same isothermal treatment and presented a degradation rate of 0.03 ± 0.003% per min.
In view of the results obtained from TGA analysis, it can be clearly concluded that the surface modification of BG particles with PDA is a promising strategy to improve the thermal stability of BG filled PLLA composites. The PDA layer on the surface of BG particles may impede the degradation reaction between the Si–O− groups present on the surface of the particles and the CO groups in the polymer backbone. As a result, those samples containing mBG particles showed Tonset and Tpeak values much closer to those of pristine PLLA and at the same time are able to maintain their initial mass during an isothermal treatment at 210 °C.
3.3. Mechanical properties of PLLA/BG and PLLA/mBG composites
For the mechanical characterization of the composites, PLLA sheets containing 5, 10 or 15 vol% of BG or mBG particles were manufactured by hot-pressing and subsequently, dumbbell-shaped samples were punched out from these sheets. After hot-pressing, samples containing BG particles had a yellowish color (Fig. S5†) and were brittle, indicating a possible degradation process of the samples during the thermoplastic processing. Thus, their molecular weight was analyzed before (Mn0) and after the hot-pressing (Mn). As can be seen in Table 3, pristine PLLA did not suffer any significant loss of molecular weight during the processing. In contrast, those samples containing 5, 10 and 15 vol% of BG particles lost, respectively, 25, 63 and 83% of their initial molecular weight. As demonstrated in the previous section, modification of particles with PDA brought about an improved thermal stability in the composites. Accordingly, those samples containing 5, 10 and 15 vol% of mBG particles only lost 7, 9 and 14% of their initial molecular weight.| Sample | E (MPa) | σ u (MPa) | ε u (%) | M n/Mn0 |
|---|---|---|---|---|
| PLLA | 1558.2 ± 109.9 | 47.7 ± 5.0 | 3.9 ± 0.5 | 0.99 ± 0.05 |
| PLLA 5BG | 1569.7 ± 81.6 | 28.7 ± 3.3 | 2.0 ± 0.3 | 0.75 ± 0.01 |
| PLLA 5mBG | 1560.7 ± 84.5 | 41.3 ± 1.3 | 2.9 ± 0.3 | 0.93 ± 0.03 |
| PLLA 10BG | 1687.8 ± 229.7 | 8.6 ± 2.8 | 0.5 ± 0.2 | 0.37 ± 0.01 |
| PLLA 10mBG | 1687.6 ± 109.0 | 39.4 ± 3.3 | 2.5 ± 0.2 | 0.91 ± 0.01 |
| PLLA 15BG | — | — | — | 0.17 ± 0.03 |
| PLLA 15mBG | 1703.7 ± 119.3 | 37.9 ± 4.1 | 1.9 ± 0.2 | 0.86 ± 0.03 |
The sharp decrease in the molecular weight of those samples containing BG particles had a detrimental effect on the final mechanical properties of the composites (Fig. S6† + Table 3). Pristine PLLA is a stiff and rigid polymer showing high Young's modulus (1558.2 ± 109.9 MPa), high tensile strength (47.7 ± 5.0 MPa) but low elongation at break (3.9 ± 0.5 MPa). When 5 or 10 vol% of BG particles were incorporated, the Young's modulus increased slightly, due to the presence of stiff inorganic fillers within the soft polymer matrix. However, the samples became too brittle and as a result the ultimate tensile strength was clearly reduced with respect to pristine PLLA. For example, PLLA filled with 10 vol% of BG particles (PLLA 10BG) showed an elongation at break and ultimate tensile strength of 0.5 ± 0.2% and 8.6 ± 2.8 MPa, respectively. PLLA filled with 15 vol% of BG particles was so brittle that the tensile test could not be carried out. Even if some deterioration in the mechanical properties was also observed for PLLA composites containing mBG particles, these samples were able to maintain their mechanical properties much better than when non-coated BG particles were incorporated. In general, the Young's modulus of the samples steadily increased with the concentration of mBG particles, reaching 1703.7 ± 119.3 MPa for the samples containing 15 vol% of mBG particles (PLLA 15mBG). The incorporation of mBG particles also resulted in the embrittlement of the samples, so lower values of elongation at break and tensile strength were recorded as the concentration of mBG particles within the PLLA matrix increased. For example, elongation at break and tensile strength were reduced from the initial values of 3.9 ± 0.5% and 47.7 ± 5 MPa for pristine PLLA to 1.9 ± 0.2% and 37.9 ± 4.1 MPa for PLLA 15mBG respectively. The embrittlement of the samples due to the incorporation of mBG particles can be explained by, on the one hand, a slightly lower molecular weight of the PLLA matrix in those samples containing mBG particles and on the other, the incorporation of brittle glass particles within the polymer matrix which could result in poorer mechanical properties of the resulting composite.
The dynamic mechanical properties of the samples processed by hot-pressing were also analyzed in tensile mode (Fig. 4 and Table 4). Fig. 4 shows the storage modulus (E′) and the damping (tanδ = E′′/E′, where E′′ is the loss modulus) of pristine PLLA and PLLA filled with BG or mBG particles. It can easily be seen that the transition from the glassy to the rubbery state occurred at lower temperatures as the content of non-coated BG particles increased. For example, the onset temperature at which this transition occurred dropped from 50.0 °C for pristine PLLA to 43.5 °C for PLLA 15BG (Table 4). As previously mentioned, those samples containing BG particles suffered a dramatic decrease in molecular weight due to the degradation reaction between the BG particles and the polymer matrix during hot-pressing. In this particular case, PLLA showed a Mw of 160 × 103 g mol−1 whereas PLLA 15BG had a Mw of 81 × 103 g mol−1. It is well known that several properties of polymers are maintained almost constant above a critical molecular weight but sharply decrease below this value.29 The lower molecular weight of these samples meant shorter polymer chains with higher mobility and consequently the transition from glassy to rubbery state takes place at lower temperatures. The opposite situation was observed when mBG particles were incorporated. In this case, the transition from glassy to rubbery moved toward higher temperatures as the content of mBG particles increased, indicating a good interaction between the polymer matrix and the filler.30
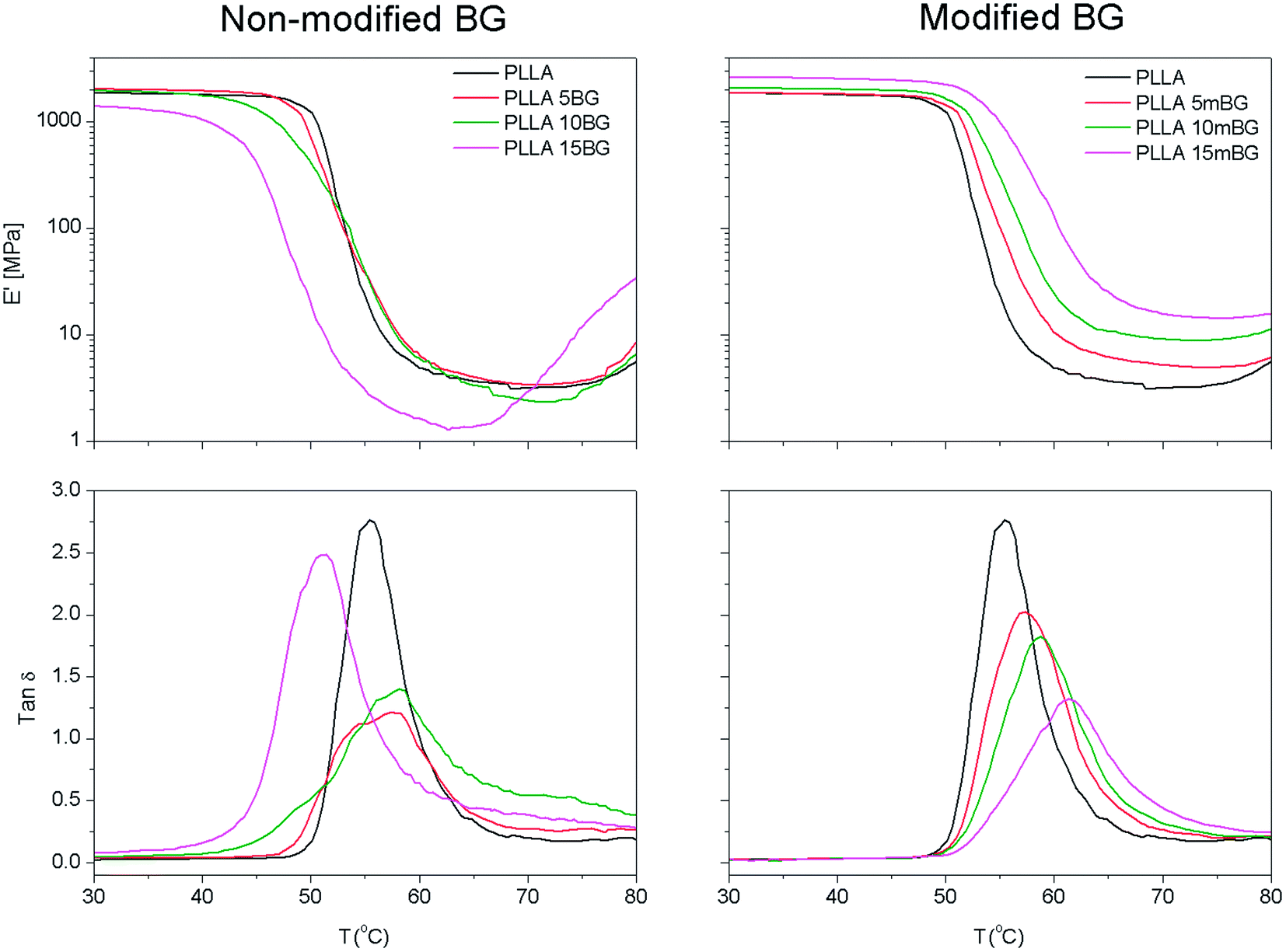 | ||
| Fig. 4
E′ and tanδ of PLLA composites containing 5, 10 or 15 vol% of BG or mBG particles. | ||
| Sample | tanδ |
T onset a (°C) | E′35 °C (MPa) | E′75 °C (MPa) | M w (g mol−1) (×103) |
|---|---|---|---|---|---|
| a Calculated as the temperature where the initial value of E′ departs from linearity. | |||||
| PLLA | 2.75 | 50.0 | 1835.6 | 3.5 | 160 |
| PLLA 5BG | 1.20 | 48.3 | 1992.9 | 3.7 | 105 |
| PLLA 5mBG | 2.19 | 50.8 | 1840.4 | 4.9 | 147 |
| PLLA 10BG | 1.40 | 47.4 | 1889.5 | 3.1 | 93 |
| PLLA 10mBG | 1.85 | 51.7 | 2047.9 | 9.0 | 149 |
| PLLA 15BG | 2.48 | 43.5 | 1303.0 | 11.8 | 81 |
| PLLA 15mBG | 1.17 | 52.9 | 2577.7 | 14.3 | 143 |
Above the transition from glassy to rubbery state, the storage modulus (E′) of PLLA, PLLA 5BG and PLLA 10BG decreased sharply and had values between 3.1 and 3.7 MPa in all cases. An increase in storage modulus was observed around 80 °C, this could be related to the cold crystallization of PLLA chains.31 This increase in E′ was more evident and occurred at lower temperatures with the PLLA 15BG sample. As previously mentioned, a sharp decrease in molecular weight was observed during hot-pressing for this sample, resulting in the formation of shorter polymer chains with higher mobility. These chains are able to reorganize themselves toward a more ordered macromolecular state at lower temperatures than samples with a higher molecular weight. In those samples containing mBG particles, the storage modulus above the glass transition (E′75 °C) was enhanced as the content of mBG particles within the composite increased, indicating a good interfacial interaction between the polymer matrix and the inorganic filler. For example, pristine PLLA had a E′75 °C of 3.5 MPa, whereas PLLA 15mBG showed a value of 14.3 MPa.
The interaction between BG particles and the PLLA matrix can also be discussed in terms of tanδ values. Composite dissipation is attributable not only to the amount of matrix present in the composite, but also to the filler–matrix interactions at the interfaces as these will tend to form layers of immobilized interphase.32 Since BG particles by themselves do not contribute to damping at the glass transition of the polymer (i.e., transition from glassy to rubbery state), composite dissipation may be given by the following equation:
| (tanδ)c/(tanδ)m = 1 − bVf | (1) |
As can be observed in Fig. 5, the results obtained for PLLA samples containing mBG particles could be fitted (R2 > 0.99) using eqn (1). The value for b was higher than 1 (b = 2.9), indicating the formation of an immobilized layer of polymer chains around mBG particles, which can be ascribed to a good filler–matrix interaction. In contrast, the values obtained for samples containing BG particles could not be fitted using eqn (1). For PLLA 5BG and PLLA 10BG samples, the (tanδ)c/(tanδ)m values were below those that correspond to b = 1. However, the (tanδ)c/(tanδ)m for the PLLA 15BG sample was higher than b = 1. This fact, together with the decrease in the temperature at which the peak of tanδ was recorded for the PLLA 15BG sample, could be associated to a plasticization effect.33,34 However, the embrittlement observed in tensile test for PLLA 15BG sample does not support this hypothesis, since a plasticization effect may have resulted in higher elongation at break for this sample. Thus, the observed higher tanδ values can be ascribed to the presence of shorter chains with higher mobility. Moreover, these short chains may possibly be located around BG particles and therefore lower the interfacial adhesion between the filler and the polymer matrix.
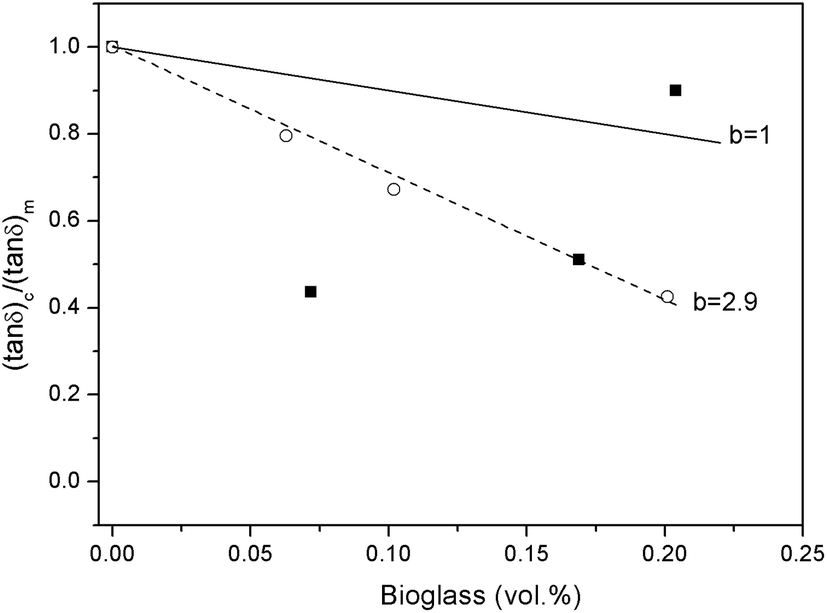 | ||
| Fig. 5 (tanδ)c/(tanδ)m values versus the BG content in the composite for samples filled with BG (■) or mBG particles (○). | ||
3.4. Bioactivity study
In the previous sections, the successful coating of BG particles with mussel-inspired PDA and the improved thermal stability of the resulting composites is highlighted. However, the main reason behind the incorporation of BG particles into synthetic polymer matrices is to impart bioactivity to the biologically inert polymer. Accordingly, in this section the bioactivity of a PLLA sample containing 15 vol% of mBG particles (PLLA 15mBG) was analyzed by submerging these samples in SBF. Fig. 6 shows SEM micrographs of the surface of PLLA 15mBG after being submerged in SBF for 28 days. Ca–P deposits were clearly observed on the surface of the sample by SEM, presenting the typical “cauliflower” morphology of hydroxyapatite. | ||
| Fig. 6 SEM micrographs of PLLA 15mBG samples submerged in SBF for 28 days. | ||
To further demonstrate the presence of hydroxyapatite on the surface of these samples, XRD analysis was performed. As can be seen in Fig. 7, peaks corresponding to crystalline hydroxyapatite were discernible in the difractogram at 32° and 26° after 28 days submerged in SBF.
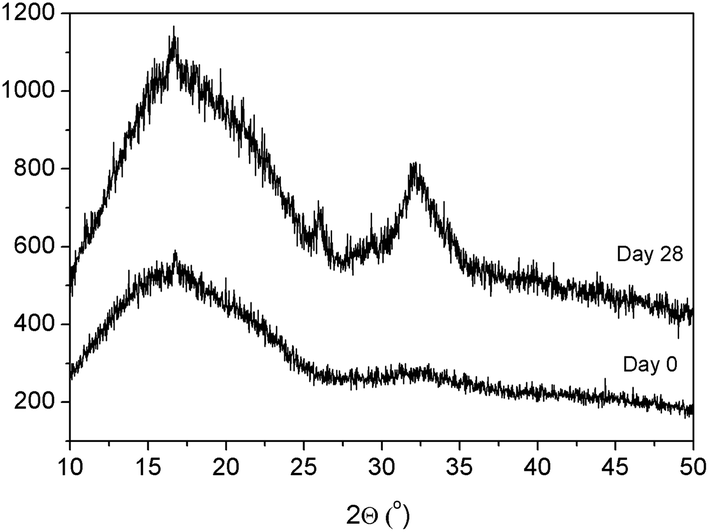 | ||
| Fig. 7 XRD patterns of PLLA 15mBG after processing and after immersion in SBF for 28 days. | ||
Moreover, mBG particles were directly immersed in SBF. At different time points (0, 1, 3 and 7 days) the FTIR spectra of these particles were recorded (Fig. S7†). The peak at 1080 cm−1 corresponds to ν3 vibration of PO43−, whereas the peak at 1458 cm−1 corresponds to ν3 vibration of CO32−. Each of these peaks increased over incubation time, representing the growth of a carbonated apatite mineral on the surface of mBG particles.
Several studies have demonstrated the enhanced deposition of hydroxyapatite layer on the surface of PDA-coated materials.35–39 The abundant catecholamine moieties provided by PDA act as a Ca2+ ion binder, facilitating the subsequent formation of hydroxyapatite crystals on the surface of the coated materials. In this study, mineralization of hydroxyapatite on the surface of PLLA samples filled with mBG particles can be ascribed to two reasons: firstly, cation (Na+ and Ca2+) exchange from mBG particles with H+ from solution, creating silanol bonds (Si–OH) forming a silica-rich network. This network facilitates the migration of Ca2+ and PO43− from the bioactive glass bulk to the surface, leading to the formation of an amorphous calcium phosphate (CaO–P2O5) rich layer. Then, by incorporation of OH− and CO32− anions from solution, the amorphous CaO–P2O5 layer is crystallized to form crystalline hydroxyapatite. Secondly, binding Ca2+ from solution with catecholamine moieties from polydopamine that can act as nucleation points for further crystalline hydroxyapatite formation.
4. Conclusion
In the present work, coating BG particles with mussel-inspired PDA is presented as a strategy to improve the thermal stability of PLLA composites. TGA analysis demonstrated that those samples filled with mBG particles showed improved thermal stability with respect to samples filled with non-coated BG particles. As a result, PLLA sheets containing mBG particles were successfully manufactured by hot-pressing, with their mechanical properties being much higher than PLLA composites filled with non-coated BG particles. DMA results indicated a good interaction between mBG particles and the PLLA matrix, as suggested by the formation of an immobilized interphase between the filler and the matrix. Finally, the bioactivity of these samples was analyzed for up to 28 days. The results obtained confirmed apatite layer formation on the surface of mBG filled PLLA samples.Acknowledgements
The authors are grateful for funds from the Basque Government, Department of Education, Universities and Research (GIC12/161-IT-632-13) and Dept. of Industry (IE10/276). A.L. thanks the University of the Basque Country (UPV-EHU) for a postdoctoral grant. SGIker technical services (UPV/EHU, MEC, GV/EJ, European Social Fund) are gratefully acknowledged for SEM, XRD and XPS support.References
- K. Rezwan, Q. Z. Chen, J. Blaker and A. R. Boccaccini, Biodegradable and Bioactive Porous Polymer/Inorganic Composite Scaffolds for Bone Tissue Engineering, Biomaterials, 2006, 27, 3413–3431 CrossRef CAS PubMed
.
- J. Fernández, A. Etxeberría and J. R. Sarasua, Synthesis, Structure and Properties of Poly(L-lactide-co-ε-caprolactone) Statistical copolymers, J. Mech. Behav. Biomed. Mater., 2012, 9, 100–112 CrossRef PubMed
- J. Fernández, A. Larrañaga, A. Etxeberría and J. R. Sarasua, Effects of Chain Microstructures and Derived Crystallization Capability on Hydrolytic Degradation of Poly(L-lactide/ε-caprolactone) Copolymers, Polym. Degrad. Stab., 2013, 98, 481–489 CrossRef
- J. Fernández, A. Larrañaga, A. Etxeberría, W. Wang and J. R. Sarasua, A New Generation of Poly(lactide/ε-caprolactone) Polymeric Biomaterials for Application in the Medical Field, J. Biomed. Mater. Res., Part A, 2014, 102, 3573–3584 CrossRef PubMed
- L. L. Hench, D. L. Wheeler and D. C. Greenspan, Molecular Control of Bioactivity in Sol–Gel Glasses, J. Sol-Gel Sci. Technol., 1998, 13, 245–250 CrossRef CAS
- I. D. Xynos, A. J. Edgar, L. D. K. Buttery, L. L. Hench and J. M. Polak, Gene-Expression Profiling of Human Osteoblasts Following Treatment with the Ionic Products of Bioglass 45S5 Dissolution, J. Biomed. Mater. Res., 2001, 55, 151–157 CrossRef CAS PubMed
- A. Larrañaga, A. Alonso-Varona, T. Palomares, E. Rubio-Azpeitia, P. Aldazabal, F. J. Martin and J. R. Sarasua, Effect of Bioactive Glass Particles on Osteogenic Differentiation of Adipose-Derived Mesenchymal Stem Cells Seeded on Lactide and Caprolactone Based Scaffolds, J. Biomed. Mater. Res., Part A, 2015 DOI:10.1002/jbm.a.35525
- V. Miguez-Pacheco, L. L. Hench and A. R. Boccaccini, Bioactive Glasses Beyond Bone and Teeth: Emerging Applications in Contact with Soft Tissues, Acta Biomater., 2015, 13, 1–15 CrossRef CAS PubMed
- L. C. Gerhardt, K. L. Widdows, M. M. Erol, C. W. Burch, J. A. Sanz-Herrera, I. Ochoa, R. Stämpfli, I. S. Roqan, S. Gabe, T. Ansari and A. R. Boccaccini, The Pro-Angiogenic Properties of Multi-Functional Bioactive Glass Composite Scaffolds, Biomaterials, 2011, 32, 4096–4108 CrossRef CAS PubMed
- A. P. Gupta and V. Kumar, New Emerging Trends in Synthetic Biodegradable Polymers-Polylactide: A Critique, Eur. Polym. J., 2007, 43, 4053–4074 CrossRef CAS
- J. Blaker, V. Maquet, R. Jérome, A. R. Boccaccini and S. N. Nazhat, Mechanical Properties of Highly Porous PDLLA/Bioglass Composite Foams as Scaffolds for Bone Tissue Engineering, Acta Biomater., 2005, 1, 643–652 CrossRef CAS PubMed
- J. Blaker, A. Bismarck, A. R. Boccaccini, A. M. Young and S. N. Nazhat, Premature Degradation of Poly(α-hydroxyesters) During Thermal Processing of Bioglass®-Containing Composites, Acta Biomater., 2010, 6, 756–762 CrossRef CAS PubMed
- A. Larrañaga and J. R. Sarasua, Effect of Bioactive Glass Particles on the Thermal Degradation Behaviour of Medical Polyesters, Polym. Degrad. Stab., 2013, 98, 751–758 CrossRef
- T. Niemela, H. Niiranen, M. Kellomaki and P. Tormala, Self-Reinforced Composites of Bioabsorbable Polymer and Bioactive Glass with Different Bioactive Glass Contents. Part 1: Initial Mechanical Properties and Bioactivity, Acta Biomater., 2005, 1, 235–242 CrossRef CAS PubMed
- H. Niiranen, T. Pyhältö, P. Rokkanen, M. Kellomäki and P. Tormala, In Vitro and In Vivo Behavior of Self-Reinforced Bioabsorbable Polymer and Self-Reinforced Bioabsorbable Polymer/Bioactive Glass Composites, J. Biomed. Mater. Res., Part A, 2004, 69, 699–708 CrossRef PubMed
- A. Larrañaga, S. Petisco and J. R. Sarasua, Improvement of Thermal Stability and Mechanical Properties of Medical Polyesters Composites by Plasma Surface Modification of the Bioactive Glass Particles, Polym. Degrad. Stab., 2013, 98, 1717–1723 CrossRef
- M. E. Lynge, R. van der Westen, A. Postma and B. Städler, Polydopamine-a Nature-Inspired Polymer Coating for Biomedical Science, Nanoscale, 2011, 3, 4916–4928 RSC
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Mussel-Inspired Surface Chemistry for Multifunctional Coatings, Science, 2007, 318, 426–431 CrossRef CAS PubMed
- H. Shen, J. Guo, H. Wang, N. Zaho and J. Xu, Bioinspired Modification of h-BN for High Thermal Conductive Composite Films with Aligned Structure, ACS Appl. Mater. Interfaces, 2015, 7, 5701–5708 CAS
- H. Wang, C. Wu, X. Liu, J. Sun, G. Xia, W. Huang and R. Song, Enhanced Mechanical and Thermal Properties of Poly(L-lactide) Nanocomposites Assisted by Polydopamine-Coated Multiwalled Carbon Nanotubes, Colloid Polym. Sci., 2014, 292, 2949–2957 CAS
- H. Wu and M. R. Kessler, Multifunctional Cyanate Ester Nancomposites Reinforced by Hexagonal Boron Nitride after Noncovalent Biomimetic Functionalization, ACS Appl. Mater. Interfaces, 2015, 7, 5915–5926 CAS
- L. Yang, S. L. Phua, C. L. Toh, L. Zhang, H. Ling, M. Chang, D. Zhou, Y. Dong and X. Lu, Polydopamine-Coated Graphene as Multifunctional Nanofillers in Polyurethane, RSC Adv., 2013, 3, 6377–6385 RSC
- L. Yang, W. A. Yee, S. L. Phua, J. Kong, H. Ding, J. W. Cheah and X. Lu, A High Throughput Method for Preparation of Highly Conductive Functionalized Graphene and Conductive Polymer Nanocomposites, RSC Adv., 2012, 2, 2208–2210 RSC
- L. Zhu, Y. Lu, Y. Wang, L. Zhang and W. Wang, Preparation and Characterization of Dopamine-Decorated Hydrophilic Carbon Black, Appl. Surf. Sci., 2012, 258, 5387–5393 CrossRef CAS
- T. Kokubo and H. Takadama, How Useful is SBF in Predicting In Vivo Bone Bioactivity?, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed
- J. Blaker, J. E. Gough, V. Maquet, I. Notingher and A. R. Boccaccini, In Vitro Evaluation of Novel Bioactive Composites Based on Bioglass-Filled Polylactide Foams for Bone Tissue Engineering Scaffolds, J. Biomed. Mater. Res., Part A, 2003, 67, 1401–1411 CrossRef CAS PubMed
- M. R. Filgueiras, G. Latorre and L. L. Hench, Solution effects on the surface-reactions of a bioactive glass, J. Biomed. Mater. Res., 1993, 27, 445–453 CrossRef CAS PubMed
- R. A. Zangmeister, T. A. Morris and M. J. Tarlov, Characterization of Polydopamine Thin Films Deposited at Short Times by Autoxidation of Dopamine, Langmuir, 2013, 29, 8619–8628 CrossRef CAS PubMed
- G. Perego, G. D. Cella and C. Bastioli, Effect of Molecular Weight and Crystallinity on Poly(Lactic Acid) Mechanical Properties, J. Appl. Polym. Sci., 1996, 59, 37–43 CrossRef CAS
- H. Lönnberg, K. Larsson, T. Lindström, A. Hult and E. Malmström, Synthesis of Polycaprolactone-Grafted Microfibrillated Cellulose for Use in Novel Bionanocomposites-Influence of the Graft Length on the Mechanical Properties, ACS Appl. Mater. Interfaces, 2011, 3, 1426–1433 Search PubMed
- J. M. Ugartemendia, A. Larrañaga, H. Amestoy and J. R. Sarasua, Supramolecular Evolution Overa n Initial Period of Biodegradation of Lactide and Caprolactone Based Medical (Co)polyesters, Polym. Degrad. Stab., 2014, 198, 87–96 CrossRef
- J. R. Sarasua and J. Pouyet, Dynamic Mechanical Behavior and Interphase Adhesion of Thermoplastic (PEEK, PES) Short Fiber Composites, J. Thermoplast. Compos. Mater., 1998, 11, 2–21 CrossRef CAS
- N. Ljungberg and B. Wesslén, Tributyl Citrate Oligomers as Plasticizers for Poly(lactic acid): Thermo-Mechanical Films Properties and Aging, Polymer, 2003, 44, 7679–7688 CrossRef CAS
- O. Martin and L. Avérous, Poly(lactic acid): Plasticization and Properties of Biodegradable Multiphase Systems, Polymer, 2001, 42, 6209–6219 CrossRef CAS
- J. Ryu, S. H. Ku, H. Lee and C. B. Park, Mussel-Inspired Polydopamine Coating as a Universal Route to Hydroxyapatite Crystallization, Adv. Funct. Mater., 2010, 20, 2132–2139 CrossRef CAS
- C. Wu, W. Fan, J. Chang and Y. Xaio, Mussel-Inspired Porous SiO2 Scaffolds with Improved Mineralization and Cytocompatibility for Drug Delivery and Bone Tissue Engineering, J. Mater. Chem., 2011, 21, 18300–18307 RSC
- J. Xie, S. Zhong, B. Ma, F. D. Shuler and C. T. Lim, Controlled Biomineralization of Electrospun Poly(ε-caprolactone) Fibers to Enhance their Mechanical Properties, Acta Biomater., 2013, 9, 5698–5707 CrossRef CAS PubMed
- M. Xu, Y. Zhang, D. Zhai, J. Chang and C. Wu, Mussel-Inspired Bioactive Ceramics with Improved Bioactivity, Cell Proliferation, Differentiation and Bone-Related Gene Expression of MC3T3 Cells, Biomaterials Sciences, 2013, 1, 933–941 RSC
- P. Yan, J. Wang, L. Wang, B. Liu, Z. Lei and S. Yang, The In Vitro Biomineralization and Cytocompatibility of Polydopamine Coated Carbon Nanotubes, Appl. Surf. Sci., 2011, 257, 4849–4855 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra09495j |
| ‡ These authors contributed to the work equally. |
| This journal is © The Royal Society of Chemistry 2015 |