
Gold nanoparticles deposited on fluorine-doped tin oxide surface as an effective platform for fabricating a highly sensitive and specific digoxin aptasensor
Abstract
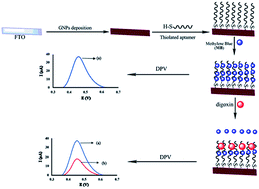
In this paper we used a modified fluorine-doped tin oxide (FTO) as the substrate of a novel aptasensor for electrochemical determination of digoxin. For this purpose, a selective thiolated digoxin aptamer was immobilized onto the gold nanoparticles deposited FTO (GNPs/FTO) surface. Cyclic voltammetry (CV), electrochemical impedance spectroscopy, scanning electron microscopy (SEM) and atomic force microscopy (AFM) were used for the characterization of modified surfaces. The aptasensor response was based on a decrease in the methylene blue (MB) current, as an electrochemical probe, after incubating with digoxin. Digoxin can be determined in a linear concentration range from 0.02 to 0.2 μg L−1 by differential pulse voltammetry (DPV). The detection limit of the proposed aptasensor was 0.01 μg L−1. Also, the fabricated aptasensor was applied to determine digoxin in urine and blood plasma samples with satisfactory results. Furthermore, high sensitivity and specificity, low detection limit and fast response, can make it possible to determine trace amounts of digoxin for routine clinical analysis.
 Please wait while we load your content...
Please wait while we load your content...

