
Electro-catalytic degradation pathway and mechanism of acetamiprid using an Er doped Ti/SnO2–Sb electrode†
Abstract
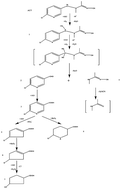
Acetamiprid, a type of new neonicotinoid pesticide, shows a high threat to water systems. Electro-catalytic degradation of acetamiprid was evaluated using an Er doped Ti/SnO2–Sb electrode prepared by the Pechini method. The acetamiprid degradation obeys first order reaction kinetics and is controlled by mass transport and oxygen evolution. TOC removal efficiency and UV scan curves revealed that some intermediate products were produced by the Er doped Ti/SnO2–Sb electrode. Through electrospray ionization quadrupole time-of-flight tandem mass spectrometry, the ion mass-to-charge ratio of intermediate products was determined. Combining the experimental results, a degradation pathway was proposed for the electro-catalytic degradation of acetamiprid. Electrodes were mainly characterized by linear sweep voltammetry and cyclic voltammetry. The acetamiprid and TOC concentrations were reduced to 87.45% and 69.31%, respectively, after 180 min of electrolysis at 10 mA cm−2.
 Please wait while we load your content...
Please wait while we load your content...

