
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A versatile approach to grafting biofouling resistant coatings from polymeric membrane surfaces using an adhesive macroinitiator
Milena
Ginic-Markovic
*a,
Thomas
Barclay
a,
Kristina T.
Constantopoulos
b,
Tawfiq
Al-Ghamdi
b,
Andrew
Blok
b,
Elda
Markovic
a and
Amanda V.
Ellis
b
aMawson Institute, University of South Australia, Mawson Lakes, South Australia 5095, Australia. E-mail: milena.ginic-markovic@unisa.edu.au; Tel: +61 8 8302 3517
bFlinders Centre for Nanoscale Science and Technology, School of Chemical & Physical Sciences, Flinders University, Sturt Road, Bedford Park, South Australia 5042, Australia
First published on 16th July 2015
Abstract
Biofouling is a serious problem for any wetted structure, having a negative influence on applications as diverse as marine transport, implanted medical devices and water treatment. Here, we address this issue by creating a polydopamine-based coating on desalination reverse osmosis membranes incorporating a bromo-macroinitiator for subsequent polymerisation of sulfobetaine monomers into anti-biofouling polymer brushes. Surface characterisation using attenuated total reflectance-Fourier transform infrared spectroscopy and the water contact angle demonstrated the attachment of the polysulfobetaine brushes and that the hydrophilicity increased for the coated membranes. Using a macroinitiator formation time of ten minutes followed by polyzwitterion coating of one hour resulted in a 17% increase in water flux without significant effect on the salt rejection performance. These membranes also exhibited substantial suppression of protein and bacterial attachment of 69% and 88% respectively compared to unmodified membranes.
Introduction
Biofouling is generated on surfaces in contact with water by the adhesion of biomacromolecules followed by the accumulation and growth of microorganisms that create a biofilm, which can ultimately allow colonisation by macrofouling organisms.1 In most instances biofouling interferes with the function of manufactured materials and consequently it is a significant problem for underwater structures such as ship hulls, implants and other biomedical devices, and also for polymer membranes used in filtration.2–4 To address surface biofouling while maintaining the bulk properties of the materials used, biofouling resistant coatings have been developed.4 Anti-biofouling coatings can incorporate biocides for the active prevention of biological growth and are often employed on manufactured surfaces in the marine environment.1,2 Hydrophobic and superhydrophobic surface coatings have also found use on marine structures, preventing adhesion of hydrophilic cells and proteins and promoting the release of any biofouling that occurs.1,4 Despite these benefits, both biocidal and hydrophobic coatings tend to lose activity over time2,5,6 and so are not ideal where reapplication of the coating is impractical. As such, many recent anti-biofouling coatings for biomedical devices are based on highly hydrophilic coatings with a neutral overall charge such as polyethylene glycol (PEG)2 and other polyethers,7 polysaccharides8 or polyzwitterions such as polybetaines3,9 and polyampholytes.10 The low interfacial energies between the surface coating and water4 combined with the formation of a hydration layer barrier at the surface11 means these surfaces exhibit low affinity both to biomacromolecules and to cells.For polymer membranes used in filtration of aqueous solutions poor hydrophilicity and fouling have been major problems affecting filtration performance,12–15 with biofouling the most widespread and difficult to address fouling problem.12,15 As such, a highly hydrophilic biofouling resistant coating would be of great benefit as it also has potential to improve flux. However, one weakness is the attachment of the coating as many approaches taken so far have significant limitations. For example, physical absorption methods result in relatively unstable coatings.13 While chemical modifications can provide a more stable coating, the use of complex and aggressive chemistries, often specific to particular membrane material and not easily scalable, means these methods are often not practical additions to the membrane manufacturing process.13 This issue can be addressed using melanin-like polydopamine (pDA), a biomimetic material developed from observation of the almost universally adhesive properties of mussel fibers.16 The dopamine monomer is deposited through self-assembly and oxidative cross-linking from mild pH aqueous solution in a rapid reaction. This results in a durable, nanoscale, hydrophilic coating that can be readily further modified as desired17,18 to introduce specific anti-biofouling measures for a particular membrane application.
For membrane filtration the increased hydrophilicity provided by an unmodified pDA surface can correlate with both increased flux and improved membrane fouling resistance in laboratory testing,19–21 although optimising the surface to achieve both together is difficult.20,22 Further, membrane filtration tests designed to mimic industrial use have revealed unmodified polydopamine coatings were not successful in reducing biofouling over extended time frames.23 As such, neutral PEG modified pDA coatings have been tried, but were also not found to reduce biofouling during long term tests,23 which may be caused by the susceptibility of PEG to oxidative degradation.10,24
Zwitterionic polymers are more oxidatively stable than PEG and have been successfully used to reduce biofouling,3,9,25–27 including analyses over extended timeframes26,28 and in application to membrane filtration.28–30 Zwitterionic coatings provide a hydrophilic, neutrally charged surface, useful to both increase flux and reduce biofouling while maintaining or enhancing salt rejection,31 but can be difficult to apply.28 Recently, the ease of application of pDA and the surface properties of zwitterionic compounds have been combined, the hybrid surface reducing biofouling and increasing flux while maintaining salt rejection for reverse osmosis polyamide membranes (PAM).19 This method utilized attachment of carboxybetaine polymers through amine groups to pDA films on membranes using a ‘grafting to’ approach.19 Concomitantly, our own research presented here has investigated a ‘grafting from’ method. In this work the PAMs were coated with a 2-bromoisobutyryl bromide (BiBBr) modified dopamine,18,32 subsequently acting to initiate polymerisation of sulfobetaine monomers from the surface using activators regenerated by electron transfer (ARGET) atom transfer radical polymerisation (ATRP) as shown in Scheme 1. This process generates polymers of controllable and narrow molecular weight distribution without the stringent experimental conditions required for other ATRP processes.33 It also enables higher polymer densities than the ‘grafting to’ approach,34 which has been identified as an important parameter in maximising the anti-biofouling properties.9 This created a hydrophilic surface that improved flux while retaining salt rejection and enabled significantly reduced biofouling by both proteins and bacteria.
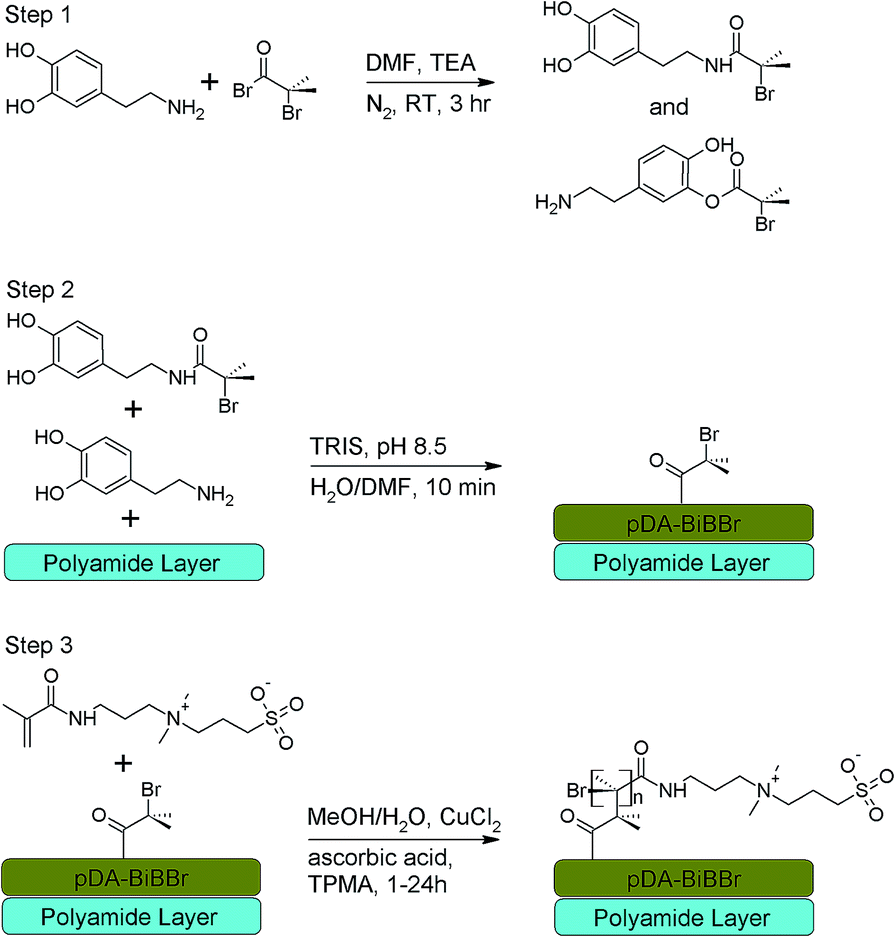 | ||
| Scheme 1 Proposed membrane modification; step 1 reaction of dopamine hydrochloride and BiBBr to give a dopamine–BiBBr initiator. Step 2 reaction of the dopamine–BiBBr initiator from step (i) in Tris buffer (pH = 8.5) to form macroinitiator coating creating PAM–pDA–BiBBr intermediate. Step 3 surface initiated ARGET-ATRP was used to graft 3SBMA from the pDA–BiBBr initiator coating to produce a pDA-g-p3SBMA modified PAM. | ||
Experimental
Materials
Anhydrous N,N'-dimethylformamide (DMF), ascorbic acid, 2-bromoisobutyryl bromide (BiBBr), copper(II) chloride (97%), dopamine hydrochloride, ethanol, glutaraldehyde solution (25% w/v in water), hydrochloric acid (37%), paraformaldehyde, phosphate buffered saline tablets, [3-(methacryloylamino)propyl]dimethyl(3-sulfopropyl) ammonium hydroxide inner salt (3SBMA), sodium acetate, sodium chloride, sodium nitrate, sodium phosphate monobasic, sucrose, triethylamine, Tris(hydroxymethyl)aminomethane (Tris) and Tris(2-pyridylmethyl)amine were purchased from Sigma-Aldrich, Australia and used as received. Isopropyl alcohol and methanol were purchased from Merck (USA) and bovine serum albumin (BSA) tetramethyl–rhodamine conjugate was obtained from Invitrogen Australia Pty Ltd, (Australia) and all used without further purification. Pure water was obtained from a Barnstead E-pure water purification system operating at a resistance of at least 18.0 MΩ cm.Commercially available polyamide membranes (PAM) for seawater reverse osmosis were provided by Toray Industries (Japan). Prior to use, coupons of PAMs (15 cm × 15 cm) were immersed in isopropanol for at least 30 min to remove components such as preservatives and to fully wet the pores, followed by immersion in pure water for 2 h.
Synthesis of polydopamine–initiator (pDA–BiBBr) modified PAMs
A dopamine–BiBBr solution was prepared following the procedure outlined by Zhu et al.18 Briefly, to make up a quantity of solution for a single coating, dopamine hydrochloride (400 mg, 2.10 mmol) was purged with nitrogen for 5 min before the addition of DMF (20 mL). Triethylamine (0.15 mL, 1.05 mmol) and BiBBr (0.13 mL, 1.05 mmol) were then added. The reaction was stirred for 3 h at room temperature, after which the dopamine–BiBBr initiator solution was used immediately for coating. A proposed scheme of the reaction is shown in Scheme 1 (step 1).In order to coat the polyamide surface of the PAMs, prepared membranes were sandwiched between a poly(methylmethacrylate) support plate and a neoprene rubber mat with a central hole cut out. A stainless steel ring was secured on top of the rubber frame, thus creating a well open to air at the top. Tris buffer (37 mM, 100 mL, pH 8.5) was added to the dopamine–BiBBr initiator solution and then immediately poured onto the polyamide surface of the PAM coupon. The coating apparatus was constantly agitated using a rocking platform (Ratek ERPM4) at ambient conditions. After 10 min the initiator modified membrane (PAM–pDA–BiBBr) was removed from the coating apparatus and thoroughly rinsed with deionized water. All PAM–pDA–BiBBr coupons were stored in methanol/pure water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 v/v) in brown glass containers until permeation measurements or further coating modifications. A proposed scheme of the reaction is shown in Scheme 1 (step 2). Membranes coated only with pDA were created from a solution of dopamine hydrochloride (2 mg mL−1) in Tris buffer (15 mM, 100 mL, pH 8.5) using the same coating apparatus under the same conditions and reaction time. PAM–pDA and control uncoated membranes were stored in methanol/pure water (1:10 v/v) in brown glass containers for consistency with the PAM–pDA–BiBBr samples.
:10 v/v) in brown glass containers until permeation measurements or further coating modifications. A proposed scheme of the reaction is shown in Scheme 1 (step 2). Membranes coated only with pDA were created from a solution of dopamine hydrochloride (2 mg mL−1) in Tris buffer (15 mM, 100 mL, pH 8.5) using the same coating apparatus under the same conditions and reaction time. PAM–pDA and control uncoated membranes were stored in methanol/pure water (1:10 v/v) in brown glass containers for consistency with the PAM–pDA–BiBBr samples.
Growth of p3SBMA from PAM–pDA–BiBBr using surface initiated ARGET-ATRP
A brown glass container containing a PAM–pDA–BiBBr modified membranes was charged with 3SBMA (10 g, 34.2 mmol), copper(II) chloride (0.001 g, 6.84 × 10−3 mmol) and Tris(2-pyridylmethyl)amine ligand (0.02 g, 6.84 × 10−2 mmol) in methanol/water (1:1 v/v; 155 mL). The container was sealed and the solution was stirred under nitrogen for 20 min. A solution of ascorbic acid reducing agent (0.6 g, 3.42 mmol) in methanol/water (1:1 v/v, 5 mL) was then added to the reaction mixture and polymerisation was conducted at room temperature for 1, 3, 6 or 24 h. Opening the container to air terminated the polymerisation reaction. After polymerisation, the pDA-g-p3SBMA modified PAM coupons were thoroughly washed with deionized water, and stored in methanol/pure water (1:10 v/v) until further analysis. A proposed scheme of the reaction is shown in Scheme 1 (step 3).
Attenuated total reflectance (ATR)-Fourier transform infrared (FTIR) spectroscopy
FTIR spectra were obtained using a Nicolet Nexus 8700 FTIR Spectrophotometer fitted with a ‘Smart Orbit’ ATR accessory containing a diamond crystal internal reflection element. PAM samples were placed active-face down on the ATR crystal and held in place by a clamp and the data was collected in air. For each sample 128 scans were taken at a resolution of 4 cm−1. A background of air was run before each sample set, and automatic baseline correction and scale normalisation were performed for each set of data. The data analysis was manipulated using Omnic Spectra software.Water contact angle
The hydrophilicity of the modified and unmodified PAMs was evaluated by performing water contact angle measurements in air using a water droplet (static sessile drop method), and analysed using Sinterface PAT1 software. Membrane samples (2 cm × 4 cm) were attached to a glass slide with double sided tape and placed on a horizontal platform. A water droplet (0.5 μL) was gently placed manually onto the membrane surface and an image captured by the camera. The internal angle of both sides of the water droplet was determined for 3 droplets at 3 different locations per sample, and the mean value ± SD of three different samples was reported.Membrane permeation tests
All permeation tests of modified and unmodified PAMs were conducted using a cross-flow filtration system (Sterlitech CF042, six units) with pure water and standard saline solution (NaCl, 2000 ppm) at 25 °C. A Hydracell Pump (M-03S) delivered the feed suspension through a Swagelock inline particulate filter (15 μm) to the cross-flow cells at a constant pressure of 2.75 MPa. The feed pressure was adjusted using a bypass needle valve (Swagelok) before the channel inlet and a back-pressure regulator at the channel outlet. All PAMs were cut to the size appropriate for the cross-flow units (14 cm × 8 cm; active membrane surface area for each cell = 42 cm2). Prior to all flux measurements, pure water was cycled for 1 h at >2.75 MPa in order to compact the membranes. Permeate was collected in glass beakers and weighed to determine flux. All balances were connected to a computer and weight measurements were collected every 5 min using a LabVIEW (National Instruments, USA) software program. Pure water flux (Jw) was calculated using eqn (1):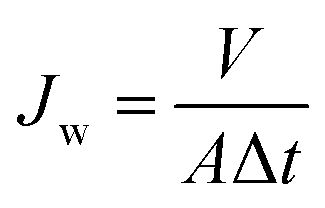 | (1) |
For salt rejection (SR) analysis, conductivities of the feed solution, concentrate and permeate were measured using a conductivity meter (Extech Equipment, Australia), and converted to concentration units (mg L−1) using a calibration curve. Salt concentration measurements (mg L−1) were used to calculate salt rejection using eqn (2):35
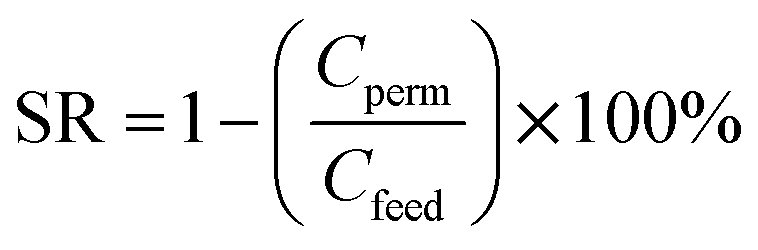 | (2) |
To eliminate the effect of the differences between each PAM, relative water flux (η) was used to characterize the variation of water flux due to coating modification type.36 The relative water flux (η) was calculated by eqn (3):
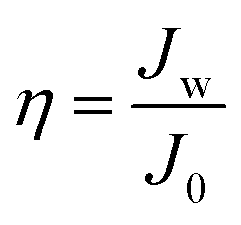 | (3) |
Similarly, relative salt rejection (τ) was used to characterize the variation of salt rejection due to coating modification type. The relative salt rejection (τ) was calculated using eqn (4):
 | (4) |
Protein absorption tests
BSA tests were performed following the procedure outlined by McCloskey et al.21 A BSA tetramethyl-tagged-rhodamine conjugate suspension was diluted to 0.1 mg mL−1 in pure water. Unmodified PAMs, pDA, pDA–BiBBr and pDA–g–p3SBMAX (where X = 1, 3, 6 or 24 h) modified PAMs were cut into 1 cm2 samples and placed into glass vials containing the BSA suspension (1 mL). The vials were kept in the dark for 1 h at room temperature. The samples were then rinsed thoroughly with pure water and allowed to dry overnight by placing the samples between two filter papers. The fluorescence intensities (I) were determined using fluorescence microscopy and analysed using Analysis 5 software. The fluorescence measured on the unmodified PAM samples immersed in pure water was designated as I0, i.e., with no BSA exposure. This background was then subtracted from the modified sample fluorescence readings.Bacteria resistance tests
The resistance of the unmodified and modified PAMs to bacterial adhesion and biofilm formation was studied by exposing the membrane samples to a biodegradable organic substrate (nutrient solution) made up of acetate, nitrate and dihydrogen phosphate in a saline solution (NaCl, 2000 ppm) to achieve a C:N:P ratio of 100:20:10.37 The nutrient solution was made by dissolving sodium chloride (2 g, 3.4 × 10−2 mol), anhydrous sodium acetate (200 mg, 2.43 × 10−3 mol), sodium nitrate (40 mg, 5.7 × 10−4 mol) and sodium phosphate monobasic (20 mg, 1.66 × 10−4 mol) in pure water (1 L). After 48 h of accelerated test, the nutrient exposed PAMs were treated with a cell fixative solution and prepared for scanning electron microscopy (SEM) imaging.
Cell-fixing solution and dehydration process
To prepare the fixative solution, paraformaldehyde (4 g) was dissolved in phosphate buffered saline (60 mL) at 60 °C. Sucrose (4 g) was then added and the solution was allowed to cool to room temperature. Glutaraldehyde solution (25% solution in water, 2 mL) was added and the final volume adjusted to 100 mL using phosphate buffered saline. Nutrient solution exposed PAM samples were gently rinsed with phosphate buffered saline (prepared from tablets dissolved in 200 mL deionized water resulting in a solution having 0.01 M phosphate, 0.137 M potassium chloride and 0.137 M sodium chloride with pH of 7.4) to remove unbound organic matter. Three replicate 1 cm2 samples per modified and unmodified PAM type were placed in vials and soaked overnight in enough fixative solution to cover the sample. After fixation for 24 h the samples were rinsed in phosphate buffered saline prior to dehydration by immersion for 15 min each in a series of aqueous ethanol baths (ethanol concentrations were 50%, 70%, 85%, 95% and 100% v/v of ethanol). The samples were then dried between filter paper overnight before preparation for SEM imaging.Scanning electron microscopy
The dried membrane samples were delaminated and mounted face-up onto SEM stubs with double-sided carbon tape. To reduce charging effects, samples were sputter-coated with platinum (5 nm) using a Cressington 208HR sputter coater, using the film thickness monitor. Samples were imaged using a FEI Quanta 450 FEG SEM. For analysis of biofouling 3 images were taken at different locations on each sample resulting in 9 images (at 10000× magnification) for each type of membrane.
Biofouling quantification
The number of bacteria was counted on all images as follows. SEM images were inspected visually, and a count was made of the number of bacteria cells per image. The mean was calculated for a minimum of 9 images – 3 images for each of 3 areas on 3 replicate sample.Statistical analysis
Statistical analyses were performed using GraphPad Prism (version 6.0; GraphPad Software, San Diego, USA). Water contact angle and bacterial count data were analysed to identify within group changes from the unmodified membrane using a one-way ANOVA followed by Tukey's post hoc test. Results were considered statistically significant if p < 0.05.Results and discussion
Optimisation and grafting of pDA-g-p3SBMA to PAMs
In previous work28,38 we demonstrated that initiator groups could be covalently linked to the PAM surface via reaction between the acid bromide of 2-bromoisobutyryl bromide and any free amine groups in the PAMs, followed by subsequent growth of p3SBMA coatings. However, this process is dependent on the availability of free amines on the membrane surface, uses aggressive organic chemistries and is not easily scalable, thereby limiting its use. Furthermore, polyamide membrane defects are reported when using direct covalent attachment of the BiBBr surface initiator.39 Therefore, we developed a method to incorporate a universal polydopamine macroinitiator coating that is independent of membrane substrate16,18,40 and easily scalable. Also, the surface attachment of oligomeric species41 using both covalent and non-covalent bonds under gentle conditions should prevent membrane defects created using standard BiBBr techniques. The pDA–BiBBr macroinitiator also has advantages compared to other similar cathecholamine adhesive bromoisobutyl initiators,42 due to the simplicity of preparation and very short deposition time.The application of the pDA-g-p3SBMA coating was achieved in a three-step process. In the first step, the amine and/or hydroxyl groups of dopamine hydrochloride were allowed to react with the BiBBr initiator,18 the mole ratio of BiBBr to dopamine hydrochloride of 0.5 statistically leaving much of the dopamine monomer unmodified to support rapid self-polymerisation.18 Subsequently, the second step was performed simply by exposing PAM surfaces to aqueous solutions of Tris buffer and dopamine–BiBBr solution, leading to the deposition of the macroinitiator. The thickness of such pDA-based coatings can be varied experimentally by varying the time of deposition,21 surpassing 10 nm in 1 h and a maximum thicknesses of 45–50 nm being achieved over 24 h.16,43 The minimum effective time of deposition was crucial as very thin layers of pDA–BiBBr are critical to avoid significant deterioration in permeation properties of membranes due to the pDA coating.16,21 Therefore, in this work the dopamine–BiBBr solution was deposited onto the active side (polyamide layer) of a membrane for 10 min to form the pDA–BiBBr macroinitiator coated PAM.
In previous work,44 this pDA–BiBBr macroinitiator was used successfully to modify a commercial PAM with subsequent surface-initiated ARGET ATRP from the surface with the anti-microbial agent, [2-(methacryloyloxy)ethyl]trimethylammonium chloride (MTAC). This positively charged monomer contains an ester bond that is relatively unstable to hydrolysis. Consequently, in this work we are using the neutrally charged and more stable 3SBMA monomer to create a coating with more general anti-biofouling application and better long-term stability. Furthermore, the zwitterionic 3SBMA has good biocompatibility,45,46 and so this material also has potential for use in biomedical applications.
Surface characterisation of the modified PAMs
ATR-FTIR measurements were performed to characterize the chemical composition of the surfaces of both unmodified and modified PAMs. The ATR-FTIR spectrum in Fig. 1a shows the typical peaks of an aromatic polyamide layer. The peaks at 1662 cm−1 and 1539 cm−1 represent the amide I (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch) and amide II (N–H bending) respectively, while the peak at 1611 cm−1 represents the CC stretching of an aromatic amine.47 In addition, peaks from the supporting polysulfone layer were also observed at 1484 cm−1, 1502 cm−1 and 1581 cm−1, a result of the depth of penetration of the IR beam using the ATR technique (∼1 μm) being greater than the thickness of the polyamide layer (∼200 nm).47
O stretch) and amide II (N–H bending) respectively, while the peak at 1611 cm−1 represents the CC stretching of an aromatic amine.47 In addition, peaks from the supporting polysulfone layer were also observed at 1484 cm−1, 1502 cm−1 and 1581 cm−1, a result of the depth of penetration of the IR beam using the ATR technique (∼1 μm) being greater than the thickness of the polyamide layer (∼200 nm).47
 | ||
| Fig. 1 ATR-FTIR spectra of (a) unmodified PAM and (b) pDA-g-p3SBMA24 modified PAM. | ||
Characteristic peaks in the spectra of pDA and pDA–BiBBr modified PAMs were not discernably different to the unmodified PAM (spectra not shown) due to the very thin layer of pDA and pDA–BiBBr (<10 nm) after 10 min of deposition.16 The successful grafting of p3SBMA from the PAM, as shown in the ATR-FTIR spectrum in Fig. 1b, provides evidence of the macroinitiator attachment. The spectrum of pDA-g-p3SBMA24 shows peaks attributable to p3SBMA at 1535 cm−1, 1481 cm−1, 1207 cm−1, and 1033 cm−1 corresponding to the N–H bending, quaternary ammonium, SO asymmetric stretching and SO symmetric stretching respectively.48
In order to evaluate the hydrophilicity of the modified and unmodified membranes, water contact angle measurements were carried out in air with a water droplet (static sessile drop method). Water contact angle measurements for the unmodified, pDA modified, pDA–BiBBr modified, pDA-g-p3SBMA1, pDA-g-p3SBMA3, pDA-g-p3SBMA6, and pDA-g-p3SBMA24 RO PAMs were measured and the results are shown in Fig. 2. Unmodified PAMs show the highest measurement of water contact angle at (39 ± 2°). pDA coatings are considered to increase the hydrophilicity of coated surfaces due to the presence of hydroxyl and amine functional groups, hence a lower contact angle (25.8 ± 1°) was observed after deposition of the pDA coating relative to the unmodified membrane surface.21 The pDA–BiBBr coating had a larger contact angle (36 ± 3°) than the pDA coating, which provides evidence of the immobilisation of the hydrophobic initiator groups.44
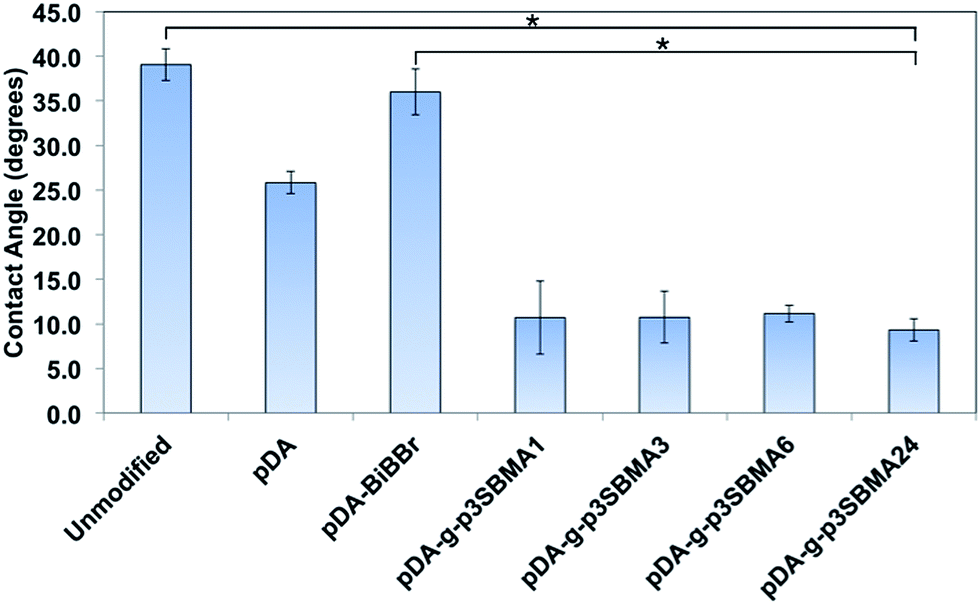 | ||
| Fig. 2 Water contact angle measurements of unmodified PAMs and modified PAMs from 1 to 24 h. Results are presented as the mean ± SD and analysed using a one-way ANOVA followed by Tukey's post ad hoc test; *represents a significant difference (p < 0.05). | ||
The increase in the hydrophilicity after grafting p3SBMA to the membrane surface for 24 h compared to both unmodified and pDA–BiBBr membranes was statistically significant (Fig. 2). However, there was no statistically significant difference between reaction times for the p3SBMA coated membranes, with all contact angles between 9 and 11°. The improved hydrophilicity of the pDA-g-p3SBMA coated PAMs was due to the formation of a hydration layer through electrostatic bonds and hydrogen bonding between the water and the coating.11 It was shown by Azzaroni et al.49 that when the thickness of a sulfobetaine layer is ≤50 nm the surface is hydrophilic (contact angle < 15°), but when the thickness is >50 nm the surface is more hydrophobic (contact angle > 40°), ascribed to conformational variations between chains of different lengths. Consequently, as all pDA-g-p3SBMA have contact angles between 9 and 11°, the thickness was less than 50 nm for all modified membranes.
Permeation properties of unmodified and modified PAMs
Modified and unmodified PAMs were analysed using a cross-flow filtration apparatus. Table 1 shows the influence of coating treatment on the fractional gain or loss of flux, which is reported as the ratio of pure water flux of modified PAMs to that of an unmodified control PAM. The pDA modified PAMs show an increase in water flux relative to the unmodified PAMs by 24% at 2.75 MPa and 10 min of deposition, which is in good agreement with reported results for similar systems.36 These increases are attributed to the increase in hydrophilicity due to the deposition of a very thin layer of pDA. Following the same logic, the slight decrease of water flux detected with a pDA–BiBBr coating relative to the unmodified PAM is as a result of the presence of hydrophobic initiator groups on the surface.| Membrane | Relative water flux (Jw/J0) | Relative salt rejection (SRcoated/SR0) |
|---|---|---|
| a The NaCl rejection (SR) for all membranes was on average ≥97.5%. | ||
| PAM | 1.00 ± 0.00 | 1.00 |
| pDA | 1.24 ± 0.19 | 0.99 |
| pDA–BiBBr | 0.90 ± 0.04 | 1.00 |
| pDA-g-p3SBMA1 | 1.17 ± 0.09 | 1.01 |
| pDA-g-p3SBMA3 | 0.74 ± 0.06 | 1.00 |
| pDA-g-p3SBMA6 | 0.73 ± 0.02 | 1.02 |
| pDA-g-p3SBMA24 | 0.79 ± 0.04 | 1.00 |
The polyzwitterionic PAM coatings led to mixed results depending on reaction time. The pDA-g-p3SBMA1 sample had increased water flux of 17% compared to the unmodified PAMs. For longer polymerisation times there was decreased water permeability from 21% to 27% relative to the unmodified PAMs, presumably due to an increase in thickness of the pSBMA coating. Despite this, all membrane coatings were still better performers than a PEG based anti-biofouling coating that reduced flux by 55%.21
The salt rejection of the unmodified and modified PAMs was measured under a transmembrane pressure of 2.75 MPa with 2000 ppm NaCl solution as the feed. Table 1 shows that the coating treatments had no significant effect on the salt rejection performance of all modified PAMs relative to the unmodified control PAM. The overall salt rejection of the modified and unmodified PAMs was >97%.
Resistance to protein adsorption and bacterial adhesion
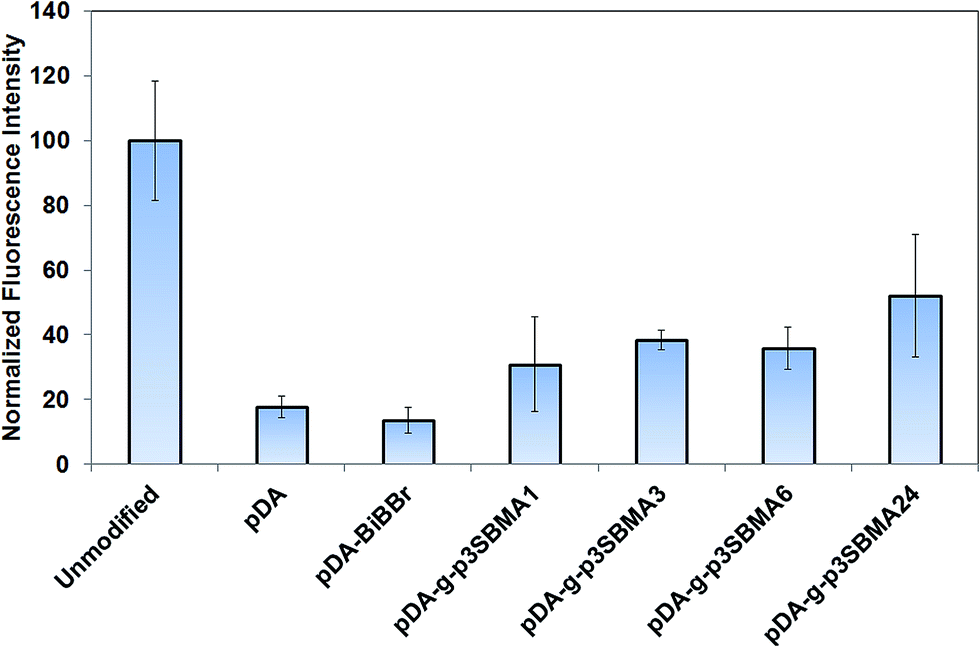 | ||
| Fig. 3 Normalized fluorescence intensity of modified and unmodified PAMs after contact with a 0.1 mg mL−1 solution of rhodamine-tagged bovine serum albumin. Values normalized to fluorescence of the unmodified PAM, which was assigned an intensity of 100. Error bars are standard deviation over 3 replicates. | ||
:N:P ratio used in the nutrient solution provides an excess dose of phosphorous, which assists to exacerbate biofouling in pure water containing naturally occurring bacteria.37 Examples of the SEM images for the unmodified, pDA, pDA–BiBBr, and p3SBMA for 1 h coated PAMs after exposure to nutrient solution are shown in Fig. 4a–d and the bacterial abundance for each sample is charted in Fig. 5. After exposing the unmodified PAMs to nutrient solution a large number of bacteria were observed on the surface (Fig. 4a). There was a significant decrease in the number of bacteria adhered to the surface for pDA and pDA–BiBBr (Fig. 4b and c), and also significant reductions in bacterial attachment observed for p3SBMA coated samples (Fig. 4d). The difference in bacterial attachment between coating times was not statistically significant.
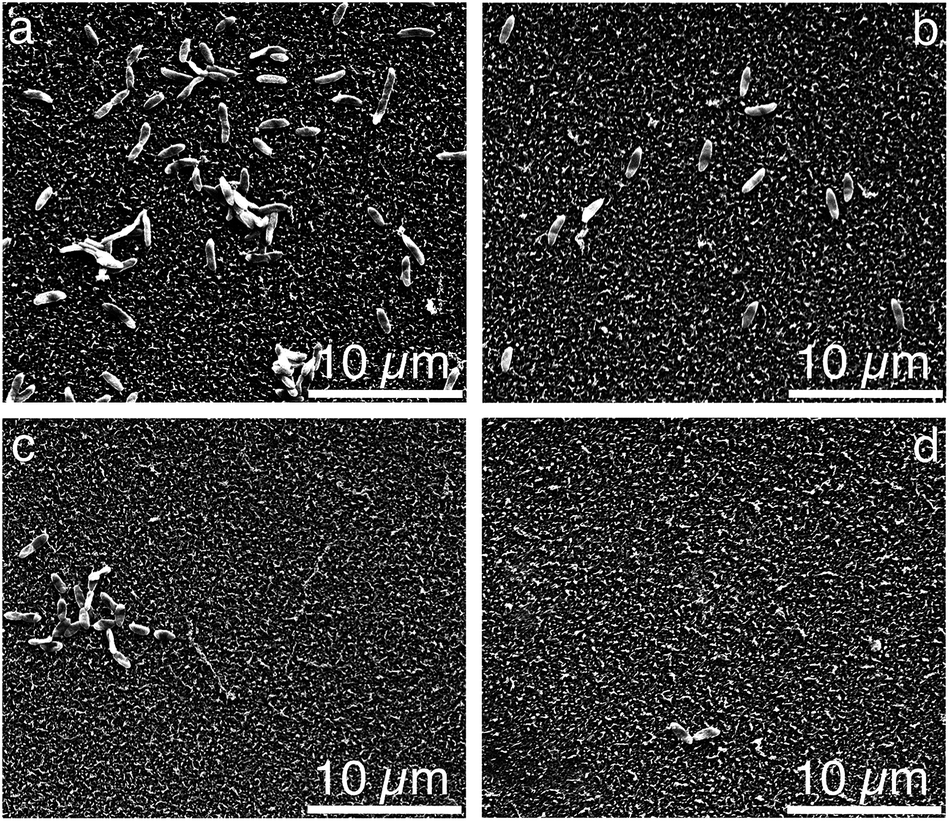 | ||
| Fig. 4 Scanning electron micrographs of (a) unmodified PAM, (b) pDA, (c) pDA–BiBBr, (d) pDA-g-p3SBMA1. | ||
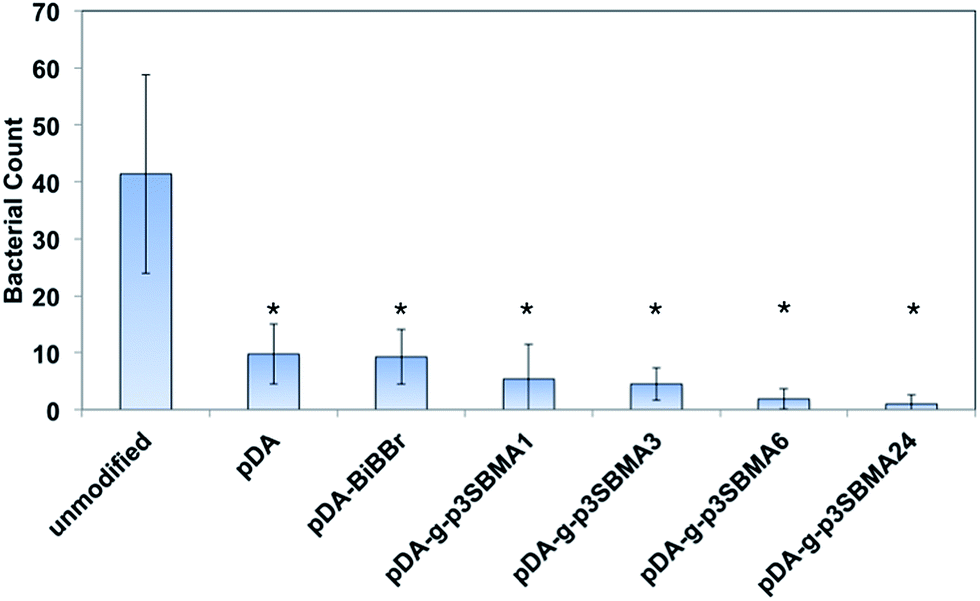 | ||
| Fig. 5 Plot of bacterial count of SEM images analysed using Image J software. Results are presented as the mean ± SD and data was analysed using a one-way ANOVA followed by Tukey's post ad hoc test; *represents a significant difference to unmodified sample (p < 0.05). | ||
For all pDA-g-p3SBMA modified PAMs a significant reduction was observed for both short-term protein adsorption and longer-term bacterial biofouling relative to unmodified PAMs (Fig. 3 and 5), in agreement with results observed for p3SBMA coated gold surfaces in the literature.45 This is because the pDA-g-p3SBMA possesses pendant side groups containing anionic sulfate and cationic quaternary ammonium groups that produce a hydration layer through electrostatic interaction in addition to hydrogen bonding. Therefore, pDA-g-p3SBMA coatings can bind a significant amount of water on the surface, as observed in the contact angle measurements (Fig. 2). This means there is a strong repulsive force preventing the hydrophobic protein and bacteria from adhering to the surface.
Conclusions
This work has demonstrated successful surface modification of commercially available SWRO PAMs with a macroinitiator followed by ARGET-ATRP of 3SBMA monomers, generating a hydrophilic, zwitterionic coating. A polymerisation time of only 1 h resulted in an increase in relative water flux of 17% over commercially available membranes, while still maintaining competitive rejection properties. Further, significant reductions in biofouling were demonstrated with protein and bacteria attachment decreased by 69% and 88% respectively for 3SBMA coated PAMs compared to unmodified PAMs. The gentle application conditions and near universal adhesive properties of the macroinitiator means that this coating has general utility in membrane filtration. Furthermore, the biocompatibility of the coating components presents opportunities in biomedical applications such as: reverse osmosis membranes for blood and kidney dialysis, drug delivery and biodevices.Acknowledgements
This work was prepared with the support and collaboration with Saudi Arabian government, Wind Prospects Pty Ltd and SA Water. Dr Dmitriy Khodakov is thanked for assistance in performing the calculation from fluorescence measurements. The authors acknowledge the financial support of the National Centre of Excellence in Desalination Australia, which is funded by the Australian Government through the Water for the Future initiative.Notes and references
- L. D. Chambers, K. R. Stokes, F. C. Walsh and R. J. K. Wood, Surf. Coat. Technol., 2006, 201, 3642–3652 CrossRef CAS PubMed
.
- I. Eshet, V. Freger, R. Kasher, M. Herzberg, J. Lei and M. Ulbricht, Biomacromolecules, 2011, 12, 2681–2685 CrossRef CAS PubMed
- Q. Liu, A. Singh and L. Liu, Biomacromolecules, 2013, 14, 226–231 CrossRef CAS PubMed
- S. Krishnan, C. J. Weinman and C. K. Ober, J. Mater. Chem., 2008, 18, 3405–3413 RSC
- X. Zhang, F. Shi, J. Niu, Y. Jiang and Z. Wang, J. Mater. Chem., 2008, 18, 621–633 RSC
- A. J. Keefe, N. D. Brault and S. Jiang, Biomacromolecules, 2012, 13, 1683–1687 CrossRef CAS PubMed
- G. Gunkel, M. Weinhart, T. Becherer, R. Haag and W. T. S. Huck, Biomacromolecules, 2011, 12, 4169–4172 CrossRef CAS PubMed
- S. Martwiset, A. E. Koh and W. Chen, Langmuir, 2006, 22, 8192–8196 CrossRef CAS PubMed
- N. D. Brault, H. S. Sundaram, Y. Li, C.-J. Huang, Q. Yu and S. Jiang, Biomacromolecules, 2012, 13, 589–593 CrossRef CAS PubMed
- T. Tah and M. T. Bernards, Colloids Surf., B, 2012, 93, 195–201 CrossRef CAS PubMed
- S. Chen, L. Li, C. Zhao and J. Zheng, Polymer, 2010, 51, 5283–5293 CrossRef CAS PubMed
- H. C. Flemming, Exp. Therm. Fluid Sci., 1997, 14, 382–391 CrossRef CAS
- G.-D. Kang and Y.-M. Cao, Water Res., 2012, 46, 584–600 CrossRef CAS PubMed
- M. Herzberg, A. Sweity, M. Brami, Y. Kaufman, V. Freger, G. Oron, S. Belfer and R. Kasher, Biomacromolecules, 2011, 12, 1169–1177 CrossRef CAS PubMed
- J. Mansouri, S. Harrisson and V. Chen, J. Mater. Chem., 2010, 20, 4567–4586 RSC
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Chem. Sci., 2013, 4, 3796–3802 RSC
- B. Zhu and S. Edmondson, Polymer, 2011, 52, 2141–2149 CrossRef CAS PubMed
- H. Karkhanechi, R. Takagi and H. Matsuyama, Desalination, 2014, 337, 23–30 CrossRef CAS PubMed
- B. D. McCloskey, H. B. Park, H. Ju, B. W. Rowe, D. J. Miller and B. D. Freeman, J. Membr. Sci., 2012, 413–414, 82–90 CrossRef CAS PubMed
- B. D. McCloskey, H. B. Park, H. Ju, B. W. Rowe, D. J. Miller, B. J. Chun, K. Kin and B. D. Freeman, Polymer, 2010, 51, 3472–3485 CrossRef CAS PubMed
- H. W. Kim, B. D. McCloskey, T. H. Choi, C. Lee, M.-J. Kim, B. D. Freeman and H. B. Park, ACS Appl. Mater. Interfaces, 2013, 5, 233–238 CAS
- D. J. Miller, P. A. Araújo, P. B. Correia, M. M. Ramsey, J. C. Kruithof, M. C. M. van Loosdrecht, B. D. Freeman, D. R. Paul, M. Whiteley and J. S. Vrouwenvelder, Water Res., 2012, 46, 3737–3753 CrossRef CAS PubMed
- S. Han, C. Kim and D. Kwon, Polymer, 1997, 38, 317–323 CrossRef CAS
- J. Cui, Y. Ju, K. Liang, H. Ejima, S. Lörcher, K. T. Gause, J. J. Richardson and F. Caruso, Soft Matter, 2014, 10, 2656 RSC
- G. Cheng, G. Li, H. Xue, S. Chen, J. D. Bryers and S. Jiang, Biomaterials, 2009, 30, 5234–5240 CrossRef CAS PubMed
- R. Lalani and L. Liu, Biomacromolecules, 2012, 13, 1853–1863 CrossRef CAS PubMed
- K. T. Constantopoulos, R. J. Pillar, E. Markovic, T. G. Barclay, K. Benkendorff, C. H. Worthley, M. Ginic-Markovic and S. R. Clarke, West Leacher, 2002, 2, 20 Search PubMed
- Y. Chiang, Y. Chang, A. Higuchi and W. Chen, J. Membr. Sci., 2009, 339, 151–159 CrossRef CAS PubMed
- R. Yang, H. Jang, R. Stocker and K. K. Gleason, Adv. Mater., 2013, 26, 1711–1718 CrossRef PubMed
- G.-R. Xu, J.-N. Wang and C.-J. Li, Desalination, 2013, 328, 83–100 CrossRef CAS PubMed
- X. Fan, L. Lin and J. L. Dalsin, J. Am. Chem. Soc., 2005, 127, 15843–15847 CrossRef CAS PubMed
- K. Matyjaszewski, H. Dong, W. Jakubowski and J. Pietrasik, Langmuir, 2007, 23, 4528–4531 CrossRef CAS PubMed
- T. Gillich, E. M. Benetti, E. Rakhmatullina, R. Konradi, W. Li, A. Zhang, A. D. Schlüter and M. Textor, J. Am. Chem. Soc., 2011, 133, 10940–10950 CrossRef CAS PubMed
- C. Bartels, R. Franks, S. Rybar, M. Schierach and M. Wilf, Desalination, 2005, 184, 185–195 CrossRef CAS PubMed
- H. Yu, Y. Cao, G. Kang, J. Liu, M. Li and Q. Yuan, J. Membr. Sci., 2009, 342, 6–13 CrossRef CAS PubMed
- J. S. Vrouwenvelder, F. Beyer, K. Dahmani, N. Hasan, G. Galjaard, J. C. Kruithof and M. C. M. Van Loosdrecht, Water Res., 2010, 44, 3454–3466 CrossRef CAS PubMed
- M. Ginic-Markovic, T. G. Barclay, K. T. Constantopoulos, E. Markovic, S. R. Clarke and J. G. Matisons, Desalination, 2015, 369, 37–45 CrossRef CAS PubMed
- Y. Zhang, Z. Wang, W. Lin, H. Sun, L. Wu and S. Chen, J. Membr. Sci., 2013, 446, 164–170 CrossRef CAS PubMed
- S. M. Kang, N. S. Hwang, J. Yeom, S. Y. Park, P. B. Messersmith, I. S. Choi, R. Langer, D. G. Anderson and H. Lee, Adv. Funct. Mater., 2012, 22, 2949–2955 CrossRef CAS PubMed
- Y. Ding, L.-T. Weng, M. Yang, Z. Yang, X. Lu, N. Huang and Y. Leng, Langmuir, 2014, 30, 12258–12269 CrossRef CAS PubMed
- J. Kuang and P. B. Messersmith, Langmuir, 2012, 28, 7258–7266 CrossRef CAS PubMed
- H. Lee, J. Rho and P. B. Messersmith, Adv. Mater., 2009, 21, 431–434 CrossRef CAS PubMed
- A. J. Blok, R. Chhasatia, J. Dilag and A. V. Ellis, J. Membr. Sci., 2014, 468, 216–223 CrossRef CAS PubMed
- W. Cho, B. Kong and I. Choi, Langmuir, 2007, 23, 5678–5682 CrossRef CAS PubMed
- J. C. Kim, M. Kim, J. Jung, H. Kim, I. J. Kim, J. R. Kim and M. Ree, Macromol. Res., 2012, 20, 746–753 CrossRef CAS
- A. Prakash Rao, S. V. Joshi, J. J. Trivedi, C. V. Devmurari and V. J. Shah, J. Membr. Sci., 2003, 211, 13–24 CrossRef
- Y. H. Zhao, K. H. Wee and R. Bai, J. Membr. Sci., 2010, 362, 326–333 CrossRef CAS PubMed
- O. Azzaroni, A. A. Brown and W. T. S. Huck, Angew. Chem., Int. Ed., 2006, 45, 1770–1774 CrossRef CAS PubMed
- M. Taylor, A. J. Urquhart, D. G. Anderson, P. M. Williams, R. Langer, M. R. Alexander and M. C. Davies, Macromol. Rapid Commun., 2008, 29, 1298–1302 CrossRef CAS PubMed
- K.-Y. Kim, E. Yang, M.-Y. Lee, K.-J. Chae, C.-M. Kim and I. S. Kim, Water Res., 2014, 54, 62–68 CrossRef CAS PubMed
- D. Bandyopadhyay, D. Prashar and Y.-Y. Luk, Langmuir, 2011, 27, 6124–6131 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2015 |