
Pharmacological evaluation of hybrid thiazolidin-4-one-1,3,5-triazines for NF-κB, biofilm and CFTR activity
Abstract
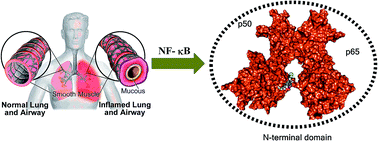
Cystic fibrosis (CF) is a monogenetic disease caused mostly by the F508del mutation, a deletion of phenylalanine at position 508 of the CF transmembrane conductance regulator (CFTR) protein, which causes improper localization and functioning of this chloride channel in lung, pancreas, and intestine by affecting the normal fluid homeostasis. In CF the lungs are the most affected organ due to the accumulation of thick mucus, which results into heavy bacterial load and associated chronic inflammation. Therefore, novel state-of-the-art therapies are needed to circumvent this problem. To address this, a series of compounds (thiazolidin-4-one-1,3,5-triazines) was tested for the inhibition of NF-κB, and compounds SP6 and SP5 showed most significant activity (respectively with relative NF-κB activity: 1.82 ± 1.87 and 1.96 ± 1.56). Docking studies of the active compounds in the DNA binding surface of the N-terminal domains of NF-κB were also carried out to identify which structural motifs are vital for activity. These compounds were also tested for antibiofilm activity against P. aeruginosa and S. aureus where they showed MIC ranges from 7.81–125 μg mL−1. The most active compound – SP6 was further assayed by micro-Ussing chamber experiments to determine its CFTR inhibitory properties, given its structural similarity to CFTR Inh172. Results suggest that SP6 does not inhibit CFTR alone or in combination with Inh172.
 Please wait while we load your content...
Please wait while we load your content...

