
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A simple and straightforward mechanochemical synthesis of the far-from-equilibrium zinc aluminate, ZnAl2O4, and its response to thermal treatment
Martin
Fabián
*ab,
Patrick
Bottke
c,
Vladimír
Girman
d,
Andre
Düvel
ef,
Klebson Lucenildo
Da Silva
ag,
Martin
Wilkening
c,
Horst
Hahn
a,
Paul
Heitjans
ef and
Vladimír
Šepelák
*abf
aInstitute of Nanotechnology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: fabianm@saske.sk; vladimir.sepelak@kit.edu; Fax: +49-721-60826368; Tel: +49-721-60828929
bInstitute of Geotechnics, Slovak Academy of Sciences, Watsonova 45, 04001 Košice, Slovakia
cInstitute for Chemistry and Technology of Materials, Graz University of Technology (NAWI Graz), Stremayrgasse 9, 8010 Graz, Austria
dInstitute of Physics, Pavol Jozef Šafárik University, Park Angelinum 9, 04154 Košice, Slovakia
eInstitute of Physical Chemistry and Electrochemistry, Leibniz University Hannover, Callinstr., 3-3a, 30167 Hannover, Germany
fCenter for Solid State Chemistry and New Materials (ZFM), Leibniz University Hannover, Callinstr., 3-3a, 30167 Hannover, Germany
gDepartment of Physics, State University of Maringá, Av. Colombo 5790, 87020-900 Maringá, Brazil
First published on 8th June 2015
Abstract
Zinc aluminate (ZnAl2O4) nanoparticles with an average size of about 10 nm are synthesized via one-step mechanochemical processing of the ZnO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :γ-Al2O3 stoichiometric mixture at ambient temperature. The mechanochemically induced formation of the phase is followed by XRD and 27Al MAS NMR. High-resolution TEM studies reveal a non-uniform nanostructure of mechanosynthesized aluminate consisting of ordered grains surrounded or separated by disordered surface and interfacial regions. Due to the capability of 27Al MAS NMR to probe the local environment of the Al cations, valuable insights into the short-range structure of ZnAl2O4 on the Ångström length scale are provided. It is demonstrated that the as-prepared aluminate possesses a partly inverse spinel structure with a far-from equilibrium arrangement of cations and distorted polyhedra, which are spatially confined to the surface and interfacial regions with a volume fraction of ca. 50% and a thickness of ca. 1 nm. The response of the nanostructured ZnAl2O4 to subsequent thermal treatment is further investigated. It turned out that the thermally induced grain growth is accompanied by a release of microstrain, by a shrinkage of the lattice parameter, as well as by a variation in the oxygen parameter and metal–oxygen bond lengths. Evidence is given of the thermally induced redistribution of cations approaching their equilibrium positions. Upon heating above 1100 K, mechanosynthesized ZnAl2O4 relaxes towards a structural state that is similar to the bulk one.
:γ-Al2O3 stoichiometric mixture at ambient temperature. The mechanochemically induced formation of the phase is followed by XRD and 27Al MAS NMR. High-resolution TEM studies reveal a non-uniform nanostructure of mechanosynthesized aluminate consisting of ordered grains surrounded or separated by disordered surface and interfacial regions. Due to the capability of 27Al MAS NMR to probe the local environment of the Al cations, valuable insights into the short-range structure of ZnAl2O4 on the Ångström length scale are provided. It is demonstrated that the as-prepared aluminate possesses a partly inverse spinel structure with a far-from equilibrium arrangement of cations and distorted polyhedra, which are spatially confined to the surface and interfacial regions with a volume fraction of ca. 50% and a thickness of ca. 1 nm. The response of the nanostructured ZnAl2O4 to subsequent thermal treatment is further investigated. It turned out that the thermally induced grain growth is accompanied by a release of microstrain, by a shrinkage of the lattice parameter, as well as by a variation in the oxygen parameter and metal–oxygen bond lengths. Evidence is given of the thermally induced redistribution of cations approaching their equilibrium positions. Upon heating above 1100 K, mechanosynthesized ZnAl2O4 relaxes towards a structural state that is similar to the bulk one.
1. Introduction
The ability of spinels to redistribute their cations over crystallographically nonequivalent positions has attracted considerable interest from many scientists. The cubic spinel structure (space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) is characterized by close-packed arrays of oxygen atoms with one eighth of the tetrahedral and one half of the octahedral sites occupied by heterovalent cations (Fig. 1). To emphasize the site occupancy on the atomic level, the structural formula of 2–3 spinels of the type M12+M223+O4 (where 2–3 refer to the valences of the M1 and M2 cations) may be written as (M11−λM2λ)[M1λM22−λ]O4, where parentheses and square brackets enclose cations that are either tetrahedrally (A) or octahedrally [B] coordinated by oxygen anions, respectively. λ represents the so-called degree of inversion that is defined as the fraction of the (A) sites occupied by trivalent (M2) cations. Spinel compounds with λ = 0 are denoted as normal spinels, whereas those with λ = 1 are called fully inverse spinels. The value of λrd = 2/3 corresponds to a random distribution of cations over the (A) and [B] positions.1 It is well recognized that physico-chemical properties of spinels are determined to a large extent by their degree of inversion.2–4 Thus, a detailed understanding of the functional behavior of spinels relies on careful characterization of their cation distribution.
m) is characterized by close-packed arrays of oxygen atoms with one eighth of the tetrahedral and one half of the octahedral sites occupied by heterovalent cations (Fig. 1). To emphasize the site occupancy on the atomic level, the structural formula of 2–3 spinels of the type M12+M223+O4 (where 2–3 refer to the valences of the M1 and M2 cations) may be written as (M11−λM2λ)[M1λM22−λ]O4, where parentheses and square brackets enclose cations that are either tetrahedrally (A) or octahedrally [B] coordinated by oxygen anions, respectively. λ represents the so-called degree of inversion that is defined as the fraction of the (A) sites occupied by trivalent (M2) cations. Spinel compounds with λ = 0 are denoted as normal spinels, whereas those with λ = 1 are called fully inverse spinels. The value of λrd = 2/3 corresponds to a random distribution of cations over the (A) and [B] positions.1 It is well recognized that physico-chemical properties of spinels are determined to a large extent by their degree of inversion.2–4 Thus, a detailed understanding of the functional behavior of spinels relies on careful characterization of their cation distribution.
 | ||
| Fig. 1 Crystal structure of normal spinel ZnAl2O4 (space group Fdm). Zn2+ and Al3+ cations are distributed over the tetrahedrally (A) and octahedrally [B] coordinated sites. | ||
In its equilibrium state, zinc aluminate (ZnAl2O4, gahnite) possesses the structure of a normal spinel (λc = 0) with the following crystal chemical formula: (Zn)[Al2]O4.5 Considerable attention has been paid to several of its multifunctional applications such as catalyst and catalyst support, UV-transparent support conductor, sensor, dielectric and optical material.6–8
The conventional solid state, i.e., ceramic, synthesis of ZnAl2O4 requires long periods of calcination of the reaction precursors at considerably high temperatures.9 In many cases, this causes the loss of zinc due to its high volatility and, consequently, it results in the formation of multiphase products and the degradation of microstructural and functional properties of the aluminate. Various wet chemistry-based routes, including, e.g., hydrothermal,10 sol–gel,11 combustion,12 co-precipitation,9 complexation,13 solvothermal6 and sonochemical14 methods, have also been developed to synthesize nanosized ZnAl2O4 powders. Most of the solution chemistry-based routes, however, still require calcination steps, at relatively low temperatures. Non-conventional mechanochemical synthesis (mechanosynthesis) has been recognized as an alternative low-temperature route; in general, it provides an efficient one-step and facile access to nanomaterials.15 In this context, the present work focuses on the one-step synthesis of nanocrystalline ZnAl2O4via mechanochemical processing of a ZnO + γ-Al2O3 mixture at ambient temperature. Although the mechanosynthesis of nanocrystalline ZnAl2O4 has already been reported in a few papers,9,16 to the best of our knowledge there is no report in the literature focusing on the defect state or the disordered local structure of ZnAl2O4 prepared by non-conventional mechanochemical routes.
Mechanosynthesized complex oxides are often inherently unstable because of their small constituent sizes, disordered structural state, and high chemical activity.17 To gain insight into thermal stability and relaxation of structural disorder, the present experimental work also deals with the study of the response of mechanosynthesized ZnAl2O4 when exposed to higher temperatures. For a comprehensive characterization of structural relaxation paths of the non-equilibrium product, we simultaneously apply X-ray diffraction (XRD), which is sensitive to medium- and long-range structural order, and 27Al magic angle spinning (MAS) nuclear magnetic resonance (NMR), which reveals local magnetic and electronic structures. Moreover, the thermally induced evolution of the aluminate synthesized is systematically monitored with Fourier transform infrared (FTIR) spectroscopy and transmission electron microscopy (TEM).
2. Experimental
Solid precursors, zinc oxide (ZnO, 99.9% purity; Aldrich) and aluminium oxide (γ-Al2O3, 99% purity; Aldrich), were used for the mechanosynthesis of ZnAl2O4. 5 g of the ZnO:γ-Al2O3 mixture was milled for various times (up to 2 h) in a high-energy planetary ball mill (Pulverisette 7 Premium line (Fritsch)). A grinding chamber (80 cm3 in volume) and balls (10 mm in diameter) made of tungsten carbide were used. The ball-to-powder weight ratio was 40:1. Milling experiments were performed in ambient atmosphere at 600 rpm. To investigate the thermally induced structural relaxation of mechanosynthesized ZnAl2O4, the material was subsequently annealed at various temperatures up to 1273 K in air for 4 hours.
In addition, polycrystalline ZnAl2O4 (with the average crystallite size ca. 105 nm) was synthesized from the mixture of ZnO and γ-Al2O3 precursors following a conventional ceramic process. This sample served as reference material. Note that an excess of ZnO (5 wt%) with respect to the stoichiometric ratio was used to avoid the formation of a multiphase product. In this case, powdered reactants were hand-milled, pressed into pellets and sintered at 1273 K for 24 hours. This process was repeated four times, reaching the final time of sintering of 120 hours.
The XRD patterns were collected using a D8 Advance diffractometer (Bruker) operating with Cu Kα radiation in Bragg–Brentano configuration. The generator was set up at 40 kV and 40 mA. The divergence and receiving slits were 0.3° and 0.1 mm, respectively. The patterns were recorded in the range of 20° to 105° 2θ with a step of 0.02° and a measuring time of 20 s. The JCPDS PDF database18 was utilized for phase identification. Rietveld refinements of XRD data of the as-prepared and subsequently annealed samples were performed using the Fullprof computer program19 utilizing regular Thompson–Cox–Hastings pseudo-Voigt profile parameters. In order to obtain proper geometry set-up and to eliminate instrumental broadening the instrumental resolution function was determined by the refinement of the LaB6 standard specimen. The cubic spinel structure of ZnAl2O4 was visualized using the Diamond program.20
The morphology of powders was studied using a combined field-emission (scanning) transmission electron microscope (S)TEM (JEOL JEM-2100F). Prior to the TEM investigations, the powders were crushed in a mortar, dispersed in ethanol, and fixed on a copper-supported carbon grid.
27Al MAS NMR measurements were performed using an Avance III 500 MHz spectrometer (Bruker) connected to an 11.4 T magnet corresponding to a Larmor frequency of 130.29 MHz for 27Al. Some of the samples were measured with an Avance III 600 MHz spectrometer (14.1 T, 156.4 MHz Larmor frequency for 27Al). At both spectrometers the samples investigated were rotated in a 2.5 mm rotor at a spinning speed of 30 kHz. Typically, 64 scans were acquired with a repetition delay of 5 s. Spectra have been referenced to aqueous Al(NO3). Since 27Al is a half-integer quadrupole nucleus (spin-quantum number I = 5/2) we used short excitation pulses close to a π/12 pulse to record spectra being useful for a quantitative analysis of site occupancies. This is especially important for Al sites with large quadrupole coupling constants. In general, a π/[4(I + 1/2)] pulse should be applied for such purpose.21 Here, the degree of inversion was estimated from the intensity ratio of the NMR lines corresponding to (A)- and [B]-site Al ions, according to the formula λ = 2I(A)/(I(A) + I[B]).
FTIR experiments were carried out using a Tensor 27 (Bruker) spectrometer. The spectra were taken in transmission mode within the range of 1200–380 cm−1.
3. Results and discussion
The mechanically induced formation of ZnAl2O4 from the ZnO:γ-Al2O3 mixture was followed by XRD (Fig. 2). After 2 hours of intensive milling, all diffraction peaks above the background are attributed to mechanosynthesized ZnAl2O4 (JCPDS PDF 82-1043).18 The broad shape of XRD reflections for mechanosynthesized aluminate, in contrast to the relatively narrow reflections for bulk ZnAl2O4 (see Fig. 2 at the bottom), provides clear evidence for a nanoscale nature of the oxide prepared via mechanosynthesis.
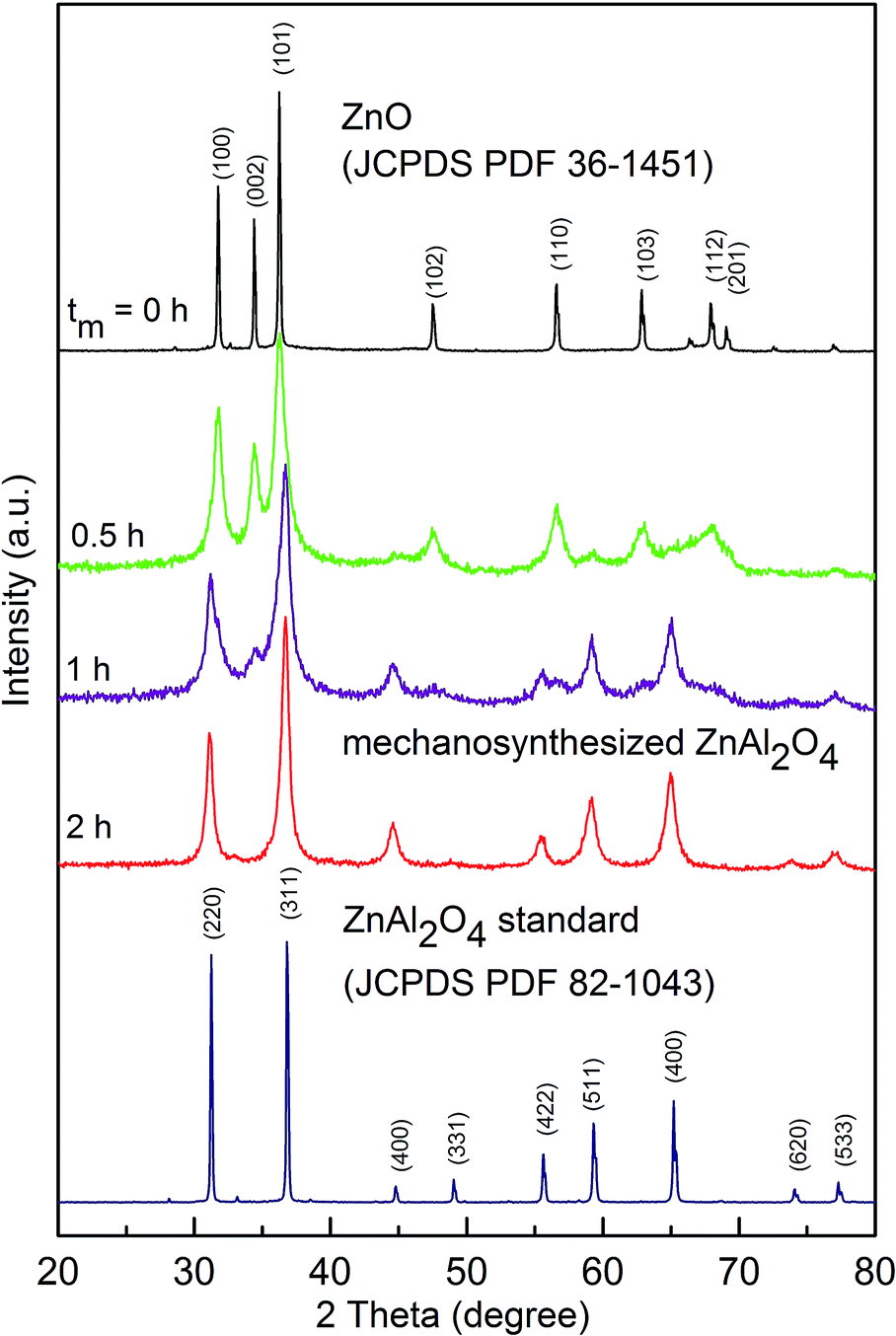 | ||
| Fig. 2 XRD patterns of the ZnO:γ-Al2O3 mixture milled for various times (up to 2 h); the milling times, tm, are shown in the figure. For comparison, bulk ZnAl2O4 prepared by a conventional ceramic route is shown at the bottom. Diffraction peaks of the ZnO precursor and bulk ZnAl2O4 are denoted by Miller indices. Note that any diffraction peaks of highly porous γ-Al2O3 are not visible due to its amorphous nature.22 | ||
A representative TEM micrograph of nanocrystalline mechanosynthesized ZnAl2O4 is shown in Fig. 3. It reveals that the aluminate consists of nanoparticles with a size distribution ranging from about 5 to 40 nm; the average crystallite size (D) is estimated to be approximately 10 nm. As shown in Fig. 3a and b, the nm-sized crystallites tend to agglomerate. They are found to be roughly spherical with a so-called core–shell structure consisting of ordered inner cores (grains) surrounded or separated by disordered surface regions (see Fig. 3c). The thickness t of the disordered surface shell estimated via high-resolution TEM was found to be about 1 nm.
 | ||
| Fig. 3 (a and b) Bright-field TEM images of nanocrystalline mechano-synthesized ZnAl2O4, (c) the core–shell configuration of mechanosynthesized nanoparticles; the thickness of the surface shell ∼1 nm is evident. The lattice fringes correspond to the crystallographic plane (220) (d = 2.86 Å) of the ZnAl2O4 phase (JCPDS PDF 82-1043). | ||
To determine the phase evolution of the ZnO:γ-Al2O3 mixture during high-energy milling in greater detail and to provide insight into the local structural disorder of the aluminate nanoparticles, the mechanochemical reaction was also followed by 27Al MAS NMR. High-resolution NMR has been proven to be highly useful to shed light on local magnetic and electric structures around the aluminium ions. In particular, this includes also local coordination and any distortions of the oxygen polyhedra.15,23Fig. 4 compares 27Al MAS NMR spectra of the as-prepared nanomaterial with that of the bulk ZnAl2O4 standard. The NMR spectrum of the bulk aluminate is dominated by a single line showing up at ca. 15 ppm which corresponds to Al ions in octahedral coordination of oxygen ions.24 This gives evidence for a normal spinel structure (λ = 0) of the aluminate, (Zn)[Al2]O4. Note that due to the interaction of the 27Al quadrupole moment with a non-vanishing electric field gradient at the Al3+[B] site, which arises from an asymmetric charge distribution, the central line is perturbed by second-order quadrupole effects.5 Such effects cannot be eliminated by magic angle spinning. The interaction manifests itself in an NMR intensity with two maxima.
 | ||
| Fig. 4 Comparison of 27Al MAS NMR spectra of the mechanosynthesized and the reference ZnAl2O4 recorded at 14.1 T and spinning speed of 30 kHz. The spectral peaks corresponding to (A)- and [B]-site Al ions are indicated in the figure. | ||
In contrast to our reference sample, the NMR spectrum of the mechanosynthesized sample consists of two well resolved and separated lines being characteristic of tetrahedrally coordinated aluminium, Al3+(A), (70 ppm) and octahedrally coordinated aluminium, Al3+[B] (18 ppm). From the intensity ratio of the (A) and [B] spectral components one can estimate the cation distribution in the material; the degree of inversion of mechanosynthesized ZnAl2O4 is found to be λ = 0.31(2). The non-equilibrium cation distribution in the nanomaterial can be characterized as follows: (Zn0.7Al0.3)[Zn0.3Al1.7]O4.
The change in cationic order in spinels is usually induced by high temperature,25 high pressure,26 irradiation of the material with electrons, ions or neutrons,27,28 and its particle size reduction to the nanometer range.5 All of these processing parameters were found to affect the cation distribution towards random arrangement (λ → λrd).15 For example, O'Neill and Dollase performed a detailed temperature-dependent structural study of ZnAl2O4.29 For samples rapidly quenched from a high temperature in the range of 973–1673 K, they found, with temperature increasing, a small increase in the degree of inversion. λ increased from 0.01 to 0.06. The extraordinary high value of λ = 0.31(2), derived for our mechanosynthesized material, demonstrates the far-from-equilibrium nature that is accessible via the mechanochemical preparation route used.
By analogy with the non-uniform configuration of the mechanochemically prepared nanooxides,15 the λ value determined for mechanosynthesized ZnAl2O4 can be considered as a mean value reflecting the cation distribution within its ordered grains and disordered interfaces and surfaces. Note that the atomic configurations in these regions of spinel oxides prepared by mechanochemical routes are chiefly characterized by a random arrangement of cations (λrd).30 In contrast, the ordered grains of nano-oxides were found to exhibit an equilibrium cation distribution (λc).30 Thus, the experimentally determined λ value for mechanosynthesized ZnAl2O4 can be expressed as λ = (1 − w)λc + wλrd, where w is the volume fraction of disordered regions. The estimated value of w = 0.465 indicates that about 50% of the atoms in the aluminate mechanosynthesized are in a structurally disordered state. Note that the simultaneous presence of two spinel phases characterized by different inversion parameters (λc and λrd) with w = 0.80 has also been observed in ZnAl2O4 irradiated with Au ions.28
Assuming a spherical shape of the as-prepared nanoparticles and taking their average diameter (D = 10 nm) as determined experimentally by TEM into account (see Fig. 3), one can deduce information on the thickness of the disordered interfacial regions in the nanomaterial [w = 1 − (1 − 2t/D)3]. The resulting t, which is 0.94 nm, is comparable to the unit cell dimension (a) of the material. We note that, in general, 1 nm is a typical thickness of grain boundaries or surface shell regions in nanostructured mechanochemically prepared oxides, such as spinels, olivines, perovskites, as well as orthorhombic and ilmenite-type complex oxides.15
In the following, we will present and discuss the results obtained when mechanosynthesized ZnAl2O4 is exposed to higher temperatures. As it is shown in Fig. 5, subsequent annealing of ZnAl2O4 results in significant narrowing of the diffraction peaks, indicating recrystallization of the sample accompanied by crystallite growth and a release of accumulated microstrain. At temperatures above 1148 K, tiny reflections belonging to ZnO appear in the XRD pattern of the material heat treated. This is due to the thermally induced partial decomposition of the highly non-equilibrium ZnAl2O4. A similar behaviour has already been observed during thermal relaxation of mechanochemically treated ZnFe2O4.31
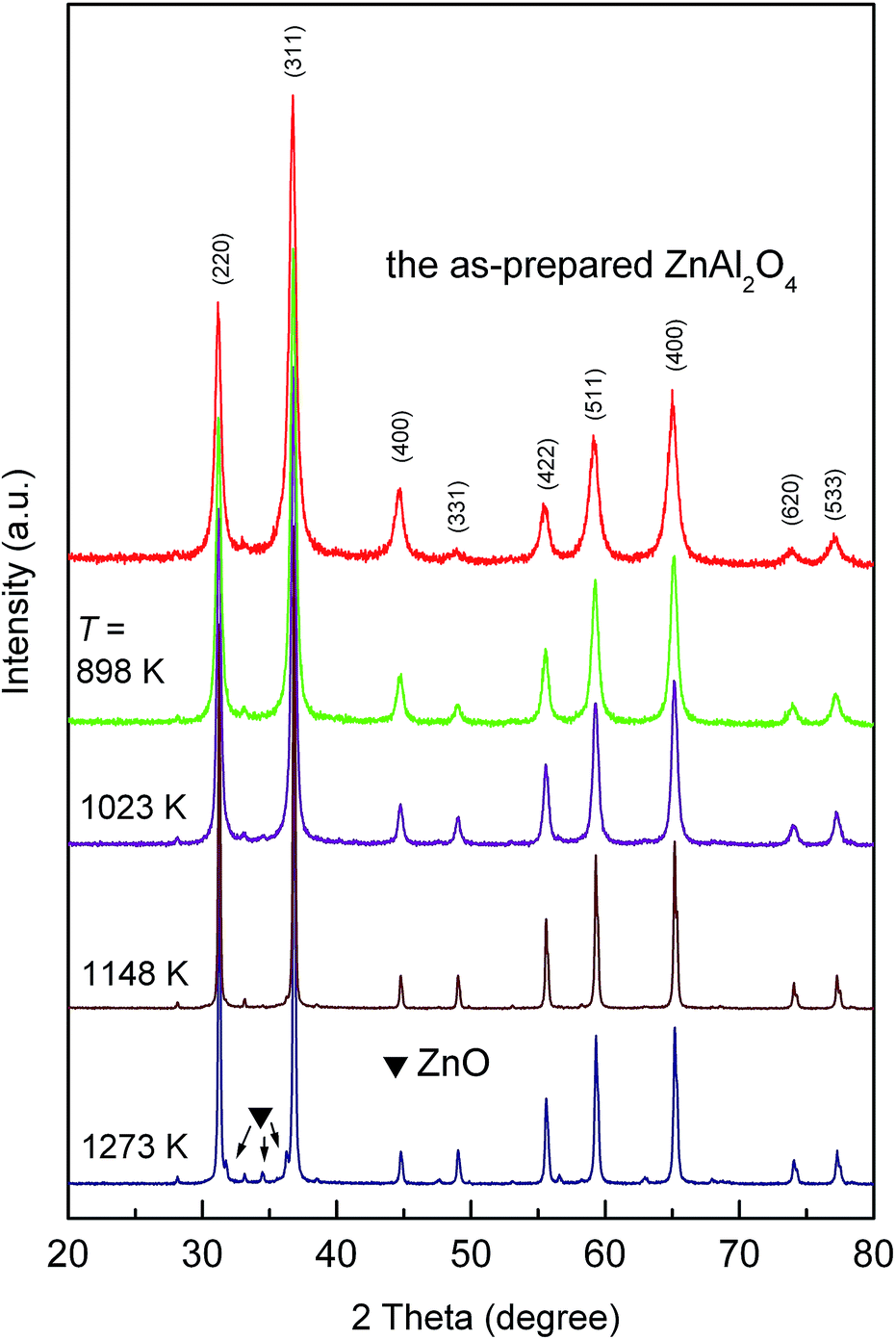 | ||
| Fig. 5 XRD patterns of the as-prepared ZnAl2O4 taken after the material has been annealed for 4 hours at the temperatures indicated. | ||
Rietveld analyses of the XRD data of the samples thermally treated enabled us to quantitatively characterize their thermally induced evolution. As is seen in Fig. 6a, the lattice parameter of the mechanosynthesized material (a = 8.121 Å) was found to be larger than that of polycrystalline ZnAl2O4 (a = 8.088 Å), which served as a reference here. The disappearance observed for the lattice expansion with increasing annealing temperature can be ascribed to a structural relaxation of the sample towards its equilibrium structure. It was found that both the crystallite size and microstrain do not change significantly with annealing temperature up to about 900 K (see Fig. 6b and c). The two properties, however, alter considerably at temperatures ranging from 900 K to 1273 K. Annealing also leads to a recovery of the local structure of the aluminate; Fig. 6d shows that the degree of inversion decreases with annealing temperature; λ changes from 0.34(2) to about 0.02(2) after treatment at 1273 K. The relaxation of the cation distribution towards its equilibrium state is accompanied by changes in the geometry of the structural units of the material. Fig. 6e shows the thermally induced variations in the cation–oxygen bond lengths in tetrahedrally and octahedrally coordinated polyhedra of ZnAl2O4. One can observe the opposite effect on the geometry of the polyhedra; while the cation–oxygen bond length in the tetrahedra expands with annealing temperature, the cation–oxygen bond length in the octahedra decreases. This alteration is obvious if we take into account the different radii of Zn2+ and Al3+ ions in (A) and [B] sites; the ions migrate from their nonequilibrium sites into the equilibrium ones; r(Zn2+) = 0.60 Å, r[Zn2+] = 0.74 Å, r(Al3+) = 0.39 Å, r[Al3+] = 0.54 Å.32 Finally, the oxygen parameter u was found to increase with increasing annealing temperature as it is shown in Fig. 6f. For the structurally relaxed material, obtained after thermal treatment at 1273 K, this parameter takes a value of 0.264, which is close to that reported for well crystalline ZnAl2O4.33
 | ||
| Fig. 6 Structural relaxation of the as-prepared ZnAl2O4 as a function of the annealing temperature. (a) Lattice parameter, (b) average crystallite size, (c) microstrain, (d) degree of inversion (e) cation–oxygen bond lengths for tetrahedrally (green) and octahedrally (blue) coordinated polyhedra, and (f) oxygen parameter vs. the annealing temperature of the mechanosynthesized ZnAl2O4. | ||
The response of the mechanosynthesized aluminate to changes in temperature was also followed by 27Al MAS NMR. Fig. 7 shows the 27Al MAS NMR spectra of the mechanosynthesized oxide that were recorded after heat treatment at the various temperatures indicated. Annealing the sample at temperatures of up to 523 K has no significant effect on the shape of the two NMR lines observed demonstrating a rather high stability of the product against heat treatment. At temperatures above 773 K, however, gradual crystallization of the ZnAl2O4 powders takes place. As expected, the spectral component corresponding to the Al3+(A) ions progressively vanishes because the mechanically induced inversion of the spinel structure gets lost. This is accompanied by a gradual narrowing of the NMR line shapes implying that the octahedra are increasingly less distorted after the sample has been annealed at elevated T. The shift observed for the NMR lines also suggests the formation of an ordered state that is reached after heat treatment.
 | ||
| Fig. 7 (Top) 27Al MAS NMR spectra of mechanosynthesized, as prepared ZnAl2O4 and of ZnAl2O4 samples that have been annealed at the temperatures indicated; the spectra were recorded at 11.4 T. The spectral peaks corresponding to (A)- and [B]-site Al ions are indicated. A noticeable change of chemical shifts reflects variations in the local environment of the Al nuclei; the short-dot line serves as a guide to eye. The tiny peak marked with * reveals an additional tetrahedrally coordinated Al position. (Bottom) the thermally induced growth of crystals is evident from TEM micrographs taken after treatment at 1023 K (left) and 1273 K (right). | ||
It is interesting to note that, most likely, the relaxation path involves an intermediate state with Al3+ ions located on the tetrahedral interstices 8b (see the asterisk in Fig. 7); these sites are normally not occupied by Al cations.34 Simultaneously, with increasing temperature of heat treatment, a right-hand side broadening of the profile for the Al3+[B] line (ca. 0 ppm) disappears. This broadening can be attributed to Al cations located in additional, most likely 16c octahedrally coordinated sites.34,35
The degree of inversion λ, calculated from the spectral intensities of the sample annealed at 1273 K, is approximately 0.03(3), which is well comparable with that of the reference material (λ = 0.01(3)). In detail, the results on the relaxation process of far-from-equilibrium ZnAl2O4 are listed in Table 1.
| T (K) | D (nm) | w XRD | w NMR |
|---|---|---|---|
| 298 | 10 | 0.516 | 0.465 |
| 523 | 10 | 0.501 | 0.449 |
| 773 | 13 | 0.373 | 0.408 |
| 898 | 17 | 0.296 | 0.283 |
| 1023 | 20 | 0.148 | 0.171 |
| 1148 | 57 | 0.077 | 0.119 |
| 1273 | 70 | 0.027 | 0.080 |
Furthermore, FTIR spectroscopy was employed to provide information on the relaxation process. As shown in Fig. 8, the spectrum of the as-prepared aluminate is dominated by two broadened bands centred at about 695 and 537 cm−1. They can be assigned to stretching vibrations in the oxide. A shoulder at about 790 cm−1 can be related to the vibrations of Al3+(A) ions.36,37 With increasing annealing temperature the absorption bands become sharper and the peak centred at 537 cm−1 splits into two absorption maxima at ν2 = 564 cm−1 and ν3 = 504 cm−1. This can be ascribed to the relaxation of the geometry of distorted polyhedra in ZnAl2O4. Since the position of vibrational modes is rather sensitive to the chemical nature of trivalent cations, i.e., to the bonding force between a trivalent cation and an oxygen anion,38 the observed red shift indicates the redistribution of cations from their nonequilibrium sites towards the equilibrium ones. The latter is accompanied by the gradual disappearance of the shoulder at 790 cm−1.
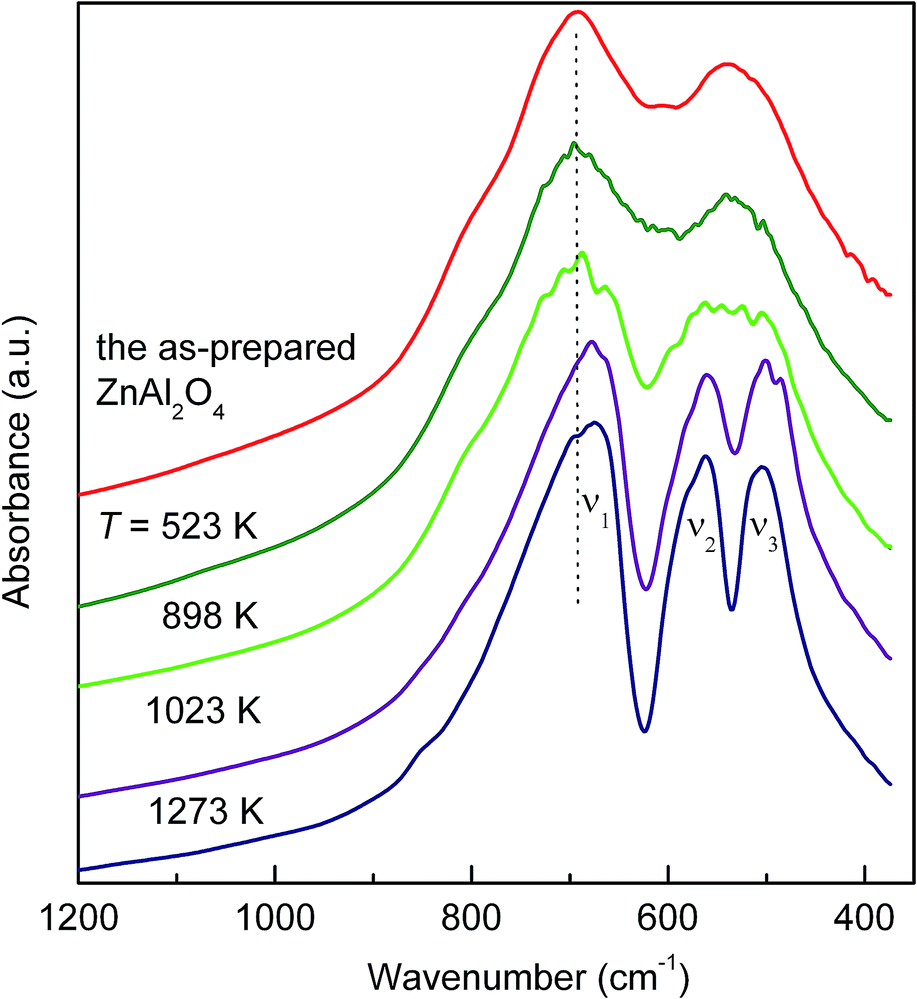 | ||
| Fig. 8 FTIR spectra of the as-prepared and annealed ZnAl2O4. The shift of the absorption bands to lower wavenumbers is ascribed to the variation in the bonding force between the trivalent cation and the oxygen anion. The shoulder observed at ca. 790 cm−1 can be related to the tetrahedrally coordinated Al3+. | ||
4. Conclusions
The present study demonstrates that nanostructured ZnAl2O4 with an average crystallite size of about 10 nm in diameter can be prepared via simple and straightforward mechanochemical synthesis starting with a stoichiometric mixture of ZnO:γ-Al2O3. The synthesis was carried out at ambient temperature and the reaction time was relatively short (2 h). It has been found that the as-prepared, nanostructured aluminate consists of ordered crystalline grains surrounded or separated by disordered interfacial regions characterized by a volume fraction of about 50%. 27Al MAS NMR spectroscopy demonstrates that the nano-aluminate is characterized by distorted polyhedra; moreover, the oxide shows a far-from equilibrium arrangement of cations characterized by degree of inversion of λ = 0.31(2). Fortunately, the range of thermal stability of the mechanosynthesized product extends up to ca. 523 K. Upon annealing at T > 773 K, the nonequilibrium cation distribution relaxes towards the equilibrium configuration. Simultaneously, the crystallites grow and the accumulated microstrain releases during annealing. This relaxation process is accompanied by a disappearance of the lattice expansion and variations in the cation–oxygen bond lengths. Thus, during heating, mechanosynthesized ZnAl2O4 relaxes towards a structural state that is similar to that of the bulk oxide.
Acknowledgements
The support by the DFG within the framework of the Priority Program “Crystalline Nonequilibrium Phases” (SPP 1415, grants no.: WI 3600 5-2 and HE 1574 11-2) and the VEGA (projects 2/0097/13 and 2/0097/14) is gratefully acknowledged. M.F. thanks “Action Austria-Slovakia” cooperation programme for support of his work at TU Graz. V.Š. acknowledges additional support by the APVV (0528-11).Notes and references
- V. Šepelák, S. M. Becker, I. Bergmann, S. Indris, M. Scheuermann, A. Feldhoff, C. Kübel, M. Bruns, N. Stürzl, A. S. Ulrich, M. Ghafari, H. Hahn, C. P. Grey, K. D. Becker and P. Heitjans, J. Mater. Chem., 2012, 22, 3117 RSC.
- V. Šepelák, I. Bergmann, D. Menzel, A. Feldhoff, P. Heitjans, F. J. Litterst and K. D. Becker, J. Magn. Magn. Mater., 2007, 316, 764 CrossRef.
- J. Wu, Z. Huang, W. Zhou, C. Ouyang, Y. Hou, Y. Gao, R. Chen and J. Chu, J. Appl. Phys., 2014, 115, 113703 CrossRef.
- A. Kan, T. Moriyama, S. Takhashi and H. Ogawa, Jpn. J. Appl. Phys., 2013, 52, 09KH01 CrossRef.
- V. Šepelák, I. Bergmann, S. Indris, A. Feldhoff, H. Hahn, K. D. Becker, C. P. Grey and P. Heitjans, J. Mater. Chem., 2011, 21, 8332 RSC.
- W. Staszak, M. Zawadzki and J. Okal, J. Alloys Compd., 2010, 492, 500 CrossRef CAS.
- N. J. van der Laag, M. D. Snel, P. C. M. M. Magusin and G. de With, J. Eur. Ceram. Soc., 2004, 24, 2417 CrossRef CAS.
- X. Y. Chen, C. Ma, Z. J. Zhang and B. N. Wang, Mater. Sci. Eng., B, 2008, 151, 224 CrossRef CAS.
- A. D. Ballarini, S. A. Bocanegra, A. A. Castro, S. R. de Miquel and O. A. Scelza, Catal. Lett., 2009, 129, 293 CrossRef CAS.
- K. Sakoda and M. Hirano, Ceram. Int., 2014, 40, 15841 CrossRef CAS; H. Zhao, Y. Dong, P. Jiang, G. Wang, J. Zhang and C. Zhang, Chem. Eng. J., 2015, 260, 623 CrossRef; M. Y. Guan, D. M. Xu, Y. F. Song and Y. Guo, Sens. Actuators, B, 2013, 188, 1148 CrossRef; Z. Chen, E. Shi, W. Li, Y. Zheng, N. Wu and W. Zhong, J. Am. Ceram. Soc., 2002, 85, 2949 CrossRef.
- R. K. Sharma and G. Ranjana, Ceram. Int., 2014, 40, 3209 CrossRef CAS; M. S. Zulfakar, H. Abdullah, W. N. W. Jalal, S. Shaari and Z. Zainuddin, Adv. Mater. Res., 2014, 895, 63 CrossRef; X. Duan, D. Yuan, X. Wang and H. Xu, J. Sol-Gel Sci. Technol., 2005, 35, 221 CrossRef.
- R. Ianos, S. Borcănescu and R. Lazău, Chem. Eng. J., 2014, 240, 260 CrossRef CAS.
- C. G. Anchieta, D. Sallet, E. L. Foletto, S. S. da Silva, O. Chiavone-Filho and C. A. O. do Nascimento, Ceram. Int., 2014, 40, 4173 CrossRef CAS.
- D. P. Dutta, R. Ghildiyal and A. K. Tyagi, J. Phys. Chem. C, 2009, 113, 16954 CAS.
- V. Šepelák, A. Düvel, M. Wilkening, K.-D. Becker and P. Heitjans, Chem. Soc. Rev., 2013, 42, 7507 RSC.
- M. V. Zdujić, O. B. Milošević and L. Č. Karanović, Mater. Lett., 1992, 13, 125 CrossRef.
- K. L. Da Silva, D. Menzel, A. Feldhoff, C. Kübel, M. Bruns, A. Paesano Jr, A. Düvel, M. Wilkening, M. Ghafari, H. Hahn, F. J. Litterst, P. Heitjans, K. D. Becker and V. Šepelák, J. Phys. Chem. C, 2011, 115, 7209 CAS.
- Joint Committee on Powder Diffraction, Standards (JCPDS) Powder Diffraction File (PDF), International Centre for Diffraction Data, Newtown Square, PA, 2004.
- J. Rodriguez-Carvajal, Fullprof Program, Version 2.4.2, ILL Grenoble, Grenoble, France, 1993 Search PubMed.
- K. Brandenburg and H. Putz, Diamond—Crystal and Molecular Structure Visualization Software, Version 3.0a, Crystal Impact GbR, Bonn, Germany, 2004 Search PubMed.
- D. Fenzke, D. Freude, T. Fröhlich and J. Haase, Chem. Phys. Lett., 1984, 111, 171 CrossRef CAS; D. Massiot, C. Bessada, J. P. Coutures and F. Taullele, J. Magn. Reson., 1990, 90, 231 Search PubMed.
- L. Samain, A. Jaworski, M. Edén, D. M. Ladd, D.-K. Seo, F. J. Garcia-Garcia and U. Häussermann, J. Solid State Chem., 2014, 217, 1 CrossRef CAS.
- A. Düvel, E. Romanova, M. Sharifi, D. Freude, M. Wark, P. Heitjans and M. Wilkening, J. Phys. Chem. C, 2011, 115, 22770 Search PubMed.
- R. L. Millard, R. C. Peterson and B. K. Hunter, Am. Mineral., 1992, 77, 44 CAS.
- S. A. T. Redfern, R. J. Harrison, H. S. C. O'Neill and D. R. R. Wood, Am. Mineral., 1999, 84, 299 CrossRef CAS.
- Z. Wang, P. Lazor, S. K. Saxena and G. Artioli, J. Solid State Chem., 2002, 165, 165 CrossRef CAS.
- T. Soeda, S. Matsumura, C. Kinoshita and N. J. Zaluzec, J. Nucl. Mater., 2000, 283–287, 952 CrossRef CAS.
- G. Baldinozzi, D. Simeone, D. Gosset, M. Dollé, L. Thomé and L. Mazérolles, Nucl. Instrum. Methods Phys. Res., Sect. B, 2006, 250, 119 CrossRef CAS.
- H. S. C. O'Neill and W. A. Dollase, Phys. Chem. Miner., 1994, 20, 541 CrossRef.
- V. Šepelák, I. Bergmann, A. Feldhoff, P. Heitjans, F. Krumeich, D. Menzel, F. J. Litterst, S. J. Campbell and K. D. Becker, J. Phys. Chem. C, 2007, 111, 5026 Search PubMed.
- V. Šepelák, L. Wilde, U. Steinike and K. D. Becker, Mater. Sci. Eng., A, 2004, 375–377, 865 CrossRef.
- R. D. Shannon, Acta Crystallogr. A, 1976, 32, 751 CrossRef.
- M. Ardit, G. Cruciani and M. Dondi, Am. Mineral., 2012, 97, 1394 CrossRef CAS.
- K. E. Sickafus, J. M. Wills and N. W. Grimes, J. Am. Ceram. Soc., 1999, 82, 3279 CrossRef CAS.
- V. Sreeja, T. S. Smitha, D. Nand, T. G. Ajithkumar and P. A. Joy, J. Phys. Chem. C, 2008, 112, 14737 CAS; H. Maekawa, S. Kato, K. Kawamura and T. Yokokawa, Am. Mineral., 1997, 82, 1125 Search PubMed.
- A. A. Da Silva, A. D. S. Conçalves and M. R. Davolos, J. Sol-Gel Sci. Technol., 2009, 49, 101 CrossRef CAS.
- P. Tarte, Spectrochim. Acta, 1967, 23, 2127 CrossRef CAS.
- J. Preudhomnei and P. Tarte, Spectrochim. Acta, 1971, 27, 1817 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |