
Fabrication of a three-dimensional graphene coating for solid-phase microextraction of polycyclic aromatic hydrocarbons†
Shuaihua
Zhang
,
Zhi
Li
,
Xiumin
Yang
,
Chun
Wang
and
Zhi
Wang
*
Department of Chemistry, College of Science, Agricultural University of Hebei, Baoding 071001, China. E-mail: wangzhi@hebau.edu.cn; zhiwang2013@aliyun.com; Fax: +86-312-7521513; Tel: +86-312-7521513
First published on 5th June 2015
Abstract
A novel three-dimensional graphene (3D-G) coated fiber for solid-phase microextraction (SPME) was fabricated via a sol–gel coating method on stainless steel wires. The 3D-G was obtained by connecting graphene oxide (GO) sheets with Ca2+ and water molecules by hydrothermal treatment. Due to the π–π stacking and hydrophobic interactions between the 3D-G and the analytes, the 3D-G fibers showed high extraction efficiencies for the polycyclic aromatic hydrocarbons (PAHs). The developed method, which combined the 3D-G coated fiber-based SPME with gas chromatography-flame ionization detection, had large enhancement factors (842–2458), low limits of detection (2.0–10.0 ng L−1) and good linear range (10.0–1000.0 ng L−1) for the PAHs in water samples. Single fiber repeatability and fiber-to-fiber reproducibility were in the range of 4.7–8.8% and 6.4–11.9%, respectively. The recoveries of the analytes for the method were in the range from 76.5% to 102.6%. The fiber exhibited an excellent durability and can be reused more than 150 times without a significant loss of the extraction performance. The method was successfully applied to the determination of PAHs in water and soil samples.
1. Introduction
Solid-phase microextraction (SPME), initially developed by Arthur and Pawliszyn1 in the early 1990s, is an environmentally friendly and solvent-free sample preparation technique which integrates sampling, extraction, preconcentration and sample introduction in a single step. Because of its simplicity, high enrichment factor and ease of operation characteristics,2 SPME has been widely applied in environmental, food, pharmaceutical and biological analysis. It is especially suitable for the preconcentration and analysis of volatile and semi-volatile organic compounds based on the partitioning of the analytes between the sample and the coating phase on the fused silica fiber.2–5 In SPME, the selection of a suitable fiber coating is the key for a particular extraction. So far, several commercial SPME fibers with a variety of polymeric coatings such as non-polar polydimethylsiloxane (PDMS), carboxen/PDMS, semi-polar PDMS/divinylbenzene (PDMS/DVB), and polar polyacrylate (PA), carbowax/PDMS, polyethyleneglycol and carbowax/templated resin have been developed. However, they suffer from some disadvantages such as high cost, easy breakage and limited variety of the fibers, which have limited its wider applications.2,3,5–7 To address some of these problems, the SPME coatings with improved properties, such as enhanced extraction efficiency and high thermal, mechanical and chemical stabilities, have been explored by analytical scientists.8–10Compared with other SPME coating materials, the carbon-based nanomaterials, which have a greater surface area-to-volume ratio and thus a greater extraction capacity, have fascinated the scientific community since their discovery.4 Recently, a number of carbon-based nanomaterials have been investigated as the coating materials for SPME, including carbon nanotubes (CNTs),4,11 graphene,3,12 carbon nanofibers,13 carbon nanocones/disks14 and ordered mesoporous carbons (OMCs).15 Graphene, an intriguing single-atom-thick two dimensional (2D) carbon material, has attracted much research attention due to its several advantages in fundamental research and practical applications.16–18 The essential 2D nanostructure of the graphene displays excellent mechanical, thermal, electrical and optical properties, which makes it the most promising carbon-based nanomaterials after fullerene and CNTs.19 Graphene also exhibits a high surface area (2630 m2 g−1) and rich stacking π electrons.20 For these reasons, graphene has been explored as the SPME coatings to extract pyrethroid pesticides,3 organochlorine pesticides (OCPs),21 polycyclic aromatic hydrocarbons (PAHs),12 polybrominated diphenyl ethers (PBDEs),22,23 phenols,24 acetanilide herbicides,25 and n-alkanes19 from water and soil samples.
However, due to its high hydrophobic property, graphene generally tends to form irreversible agglomerates or even restack to form graphite through strong π–π stacking interactions under certain conditions,26 which is undesirable for its applications. This problem has been resolved through the reassembly of 2D graphene sheets into a 3D graphene which exhibits a low density, large open pores and high internal surface area.27 In the last few years, much effort has been paid to the development of the strategies for the preparation of 3D graphene and its potential applications. Chemical self-assembly is an effective and economical approach to create 3D architectures of graphene in desired sizes and shapes for various applications in electronic, optical, electro-optical devices, sensors, and so on.28 In 2010, Shi et al.29 successfully fabricated a 3D graphene hydrogel via a convenient one-step hydrothermal technique. The combination of hydrophobic and π–π interactions is the guiding force for the formation of 3D random networks between the flexible graphene sheets. When the concentration of graphene oxide (GO) solution was sufficiently high, the cross-linking through partial overlapping of the flexible graphene sheets occurred immediately after GO was reduced, and eventually enough cross-linking sites were generated to form a 3D network with pore sizes ranging from submicrometer to several micrometers.29 Furthermore, the addition of polymers, acids, small organic molecules, biomolecules, or ions, which act as cross-linkers, can induce the formation of 3D network because of the various driving forces such as hydrogen bonding, electrostatic interaction, or coordination.28 Huang et al.30 have reported the self-assembly of 3D-G using divalent ions (Ca2+, Ni2+, or Co2+) and water molecules as cross-linkers by hydrothermal treatment. After being strengthened by polyvinyl alcohol intercalation and then freeze dried, the solid 3D architecture of microporous graphene was obtained.30
In this work, a 3D-G was prepared via a one-step hydrothermal technique by using divalent ion (Ca2+) as a cross-linker. Then, the 3D-G coated SPME fiber was prepared by immobilizing the 3D-G through a sol–gel coating method onto a prior functionalized stainless steel wire which had been coated by microstructured silver layer through silver mirror reaction to enlarge its surface area.6 The as-fabricated 3D-G coated fiber was used for the headspace SPME (HS-SPME) of polycyclic aromatic hydrocarbons (PAHs) prior to GC-FID. The main experimental parameters including the extraction time and temperature, desorption temperature and time, agitation speed and salt concentration were investigated to achieve an optimum extraction efficiency. The extraction selectivity of the 3D-G coated fiber was evaluated using some PAHs, phthalate esters (PAEs) and n-alkanes as model analytes. The fiber lifetime was also evaluated. Finally, the established HS-SPME-GC method was validated and applied for the determination of PAHs in water and soil samples.
2. Experimental
2.1. Reagents
The graphite (50 mesh) used for the preparation of GO was purchased from Boaixin (Baoding, China). GO was prepared from nature graphite according to our early report31 by a modified Hummers' method. Methyltrimethoxysilane (MTMOS, 98%) and trifluoroacetic acid (TFA, 95%) was obtained from Energy Chemical (Shanghai, China). Hydroxyl terminated polydimethylsiloxane (HO-PDMS, 98%) was bought from Heowns Biochemical Technology Co. (Tianjing, China). Other chemical reagents including ethanol (95%), hydrochloric acid (HCl, 37%), nitric acid (HNO3, 65%), sodium chloride (NaCl, 99.5%), AgNO3 (99.8%), glucose (98%) and hydroxide (NaOH, 96%), all of analytical grade, were from Sinopharm Chemical Reagent Co. (Shanghai, China). HPLC grade acetone and n-hexane were obtained from Fuchen Chemical Reagent Co. (Tianjin, China). The stainless steel wires (type 304, 350 μm o. d.) used for the SPME fiber support were obtained from Shanghai Gaoge Industrial and Trading Co. (Shanghai, China).The PAHs standards (naphthalene (NAP, 99%), acenaphthene (ANE, 99%), fluorene (FLU, 99%), phenanthrene (PHE, 99%), anthracene (ANT, 99%), fluoranthene (FLA, 99%) and pyrene (PYR, 99%)) were obtained from the Institute of Agro-Environmental Protection (Tianjin, China). The PAEs standards (dimethyl phthalate (DMP, 99.5%), diethyl phthalate (DEP, 99%), dibutyl phthalate (DBP, 99.5%) and dicyclohexyl phthalate (DCHP, 98%)) were obtained from Sinopharm Chemical Reagent Co. (Shanghai, China). The n-alkanes standards (n-undecane (n-C11, 99%), n-dodecane (n-C12, 99.8%), n-tridecane (n-C13, 99%), n-tetradecane (n-C14, 99%), n-pentadecane (n-C15, 99%) and n-hexadecane (n-C16, 99.6%)) were purchased from Acros Organics (New Jersey, USA). The three mixture stock solutions for the three groups of the analytes including PAHs, PAEs and n-alkanes were individually prepared in acetone at a concentration of 1.0 μg mL−1 for each of the analytes and stored in the dark at 4 °C for further use. For the extraction experiments, the working solutions were freshly prepared in ultrapure water at concentrations of 0.1 μg L−1 for PAHs, 0.5 μg L−1 for PAEs and 1.0 μg L−1 for n-alkanes, respectively.
2.2. Apparatus and characterization
GC analyses were carried out on an Agilent 7820A series gas chromatograph (Agilent Technologies, CA, USA) equipped with a flame ionization detector (FID) and a split/splitless injector. All the separations were performed on a HP-5 capillary column (30 m × 0.25 mm i.d. × 0.25 μm film thickness) (Agilent Scientific, USA). Nitrogen of high purity (>99.999%) was used as the carrier and make-up gas at 1.2 mL min−1 and 25 mL min−1, respectively. Hydrogen and air flow rates were maintained at 30 and 400 mL min−1, respectively. The GC chromatographic conditions for PAHs and PAEs were as follows: splitless mode; injector temperature, 250 °C; detector temperature, 300 °C; the column oven temperature program: start at 60 °C for 2 min, then increased to 200 °C at 15 °C min−1, finally increased to 260 °C at 20 °C min−1, and held for 3 min. For n-alkanes, the oven temperature was programmed: initial temperature from 60 °C for 2.0 min, then increased at 15 °C min−1 to 250 °C and held for 2 min.Scanning electron microscopy (SEM) images were obtained with a Hitachi S4800 field emission electron microscope (Hitachi, Japan) operated at 5.0 kV. X-ray photoelectron spectroscopy (XPS) experiments were performed on an ESCALAB 250 XPS with monochromated Al Kα radiation (hυ = 1486.6 eV) at 45° photoelectron takeoff angle and a 500 μm beam size. IR spectra were obtained on a Rayleigh WQF-520 Fourier transform infrared (FT-IR) spectrometer (Beijing, China). A thermogravimetric (TG) analyser (Henven HCT-2, Beijing, China) was applied to evaluate the thermal stability of the coating from room temperature to 800 °C under N2 atmosphere at a heating rate of 10 °C min−1. The laboratory-made SPME fiber was put on the manual SPME fiber holder. All of the extractions were performed in 25.0 mL glass vials with Teflon-lined caps to prevent sample evaporation. A temperature-controlled magnetic stirrer, model DF-101S (High-tech. Zone Sunshine Science Instrument Co., Baoding, China) was employed for stirring the sample during the extraction.
2.3. Preparation of three-dimensional graphene
The 3D-G was fabricated following the one-step hydrothermal strategy from aqueous GO suspensions as described previously30 with some modifications. In brief, GO (100 mg) was dispersed into 50 mL of deionized water by sonication, then 5 mg CaCl2 (Ca2+/GO weight ratio of 0.05) was added into the GO solution. After the mixture was magnetically stirred for 3 h, the GO solution was transferred into a Teflon-lined stainless steel autoclave and treated hydrothermally at 150 °C for 10 h. After the autoclave was cooled down to room temperature, a black hydrogel was formed in the bottom of the autoclave. The formed graphene sheet skeletons were interconnected and supported by chemical and hydrogen bonds which were generated from the interlinkage of the water molecules, the divalent metal ions and the oxygen containing groups (see Fig. 1). After being freeze-dried, a 3D-G with porous structure was obtained. | ||
| Fig. 1 Schematic illustration of the formation of 3D-G with Ca2+ linkage. | ||
2.4. Functionalization of stainless steel wire surface
One end (1.5 cm) of the 18 cm-long stainless steel wire (o.d. 350 μm) was first washed with NaOH aqueous solution and then dipped in nitrohydrochloric acid (HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 = 3:1, v/v) for about 15 min to remove the stable oxide on its surface and corrode the end to the diameter of about 180 μm (see Fig. S1 in the ESI†). The wire was polished smoothly and then ultrasonication-washed with acetone, methanol and double-distilled water each for 6 min, respectively. After being conditioned in a desiccator at room temperature for 24 h, the wire was dipped into a reaction solution containing 0.1 mol L−1 [Ag(NH3)2]+ and 0.5 mol L−1 glucose for about 2.5 h to form a microstructured silver layer by silver mirror reaction.19 Then, the wire was taken out and rinsed with distilled water. After being dried at room temperature, a firm and porous coating of silver was formed on the surface of the stainless steel wire.
:HNO3 = 3:1, v/v) for about 15 min to remove the stable oxide on its surface and corrode the end to the diameter of about 180 μm (see Fig. S1 in the ESI†). The wire was polished smoothly and then ultrasonication-washed with acetone, methanol and double-distilled water each for 6 min, respectively. After being conditioned in a desiccator at room temperature for 24 h, the wire was dipped into a reaction solution containing 0.1 mol L−1 [Ag(NH3)2]+ and 0.5 mol L−1 glucose for about 2.5 h to form a microstructured silver layer by silver mirror reaction.19 Then, the wire was taken out and rinsed with distilled water. After being dried at room temperature, a firm and porous coating of silver was formed on the surface of the stainless steel wire.
2.5. Preparation of three-dimensional graphene coated SPME fiber
50 mg of 3D-G was suspended in 200 μL MTMOS in a 1.5 mL centrifuge tube, and then 100 μL of HO-PDMS was added. The mixture was agitated thoroughly by sonication for 30 min. Then 50 μL of TFA (an acidic catalyst) was added and the solution was sonicated for 10 min. After that, the sol solution of 3D-G coating material was formed. The functionalized stainless steel wire was perpendicularly dipped into in the sol solution, and then pulled out slowly. Later, the fiber was vertically inserted into the extra-fine powdered 3D-G material in a 1.5 mL centrifuge tube, rotated a few cycles and pulled out. The superfluous 3D-G powder on the wire was removed by rotating the wire on a piece of clean weighing paper. Next, the coated fiber was placed in an oven at 150 °C for 30 min for drying. The above process was repeated several times until the desired thickness was obtained. At last, the coated wire was dipped into the sol solution again to form a thin film of polymer which can protect the whole coating and avoid the flaking of the powder. The coating thickness obtained after three coating cycles was around 20 μm. All the prepared fibers were placed in a desiccator overnight at room temperature. For SPME use, 1.0 cm long of the coating was kept and the other remaining coatings were scratched off. Then, the coated fiber was assembled to a 5 μL microsyringe and conditioned in the GC injector at 200 °C for 1 h and 280 °C for 1 h under a stream of nitrogen prior to their use for SPME.2.6. Sample preparation
Two water samples were collected from local river and pond (Baoding, China). They were stored in the precleaned glass bottles which had been thoroughly washed with detergents, water, methanol, and ultrapure water and dried.32 The water samples were directly used for the following HS-SPME.Soil samples were collected from the two farmlands, one near a local rubbish recycling center (Soil 1) and one close to a local thermal power plant (Soil 2). For the analytical performance assessment, a 5.0 mL mixture standard acetone solution containing each of the PAHs at 0.1 μg mL−1 was added to 10.0 g of soil to give a spiked level of 50.0 ng g−1 for each of the target compounds. After being dried at room temperature, the soil samples were sieved to a particle size of 0.45 mm. The soil samples were pretreated according to Zhang et al.12 with some modifications. In brief, 10.0 g of soil was extracted with 30.0 mL of acetone for 30 min using a rotary stirrer and then centrifuged for 10 min. The extract was filtered and evaporated to dryness at 30 °C using a rotary evaporator. Afterward, the dry residue was redissolved in 500 μL of acetone, and 15.0 μL of this solution was diluted with 15.0 mL of water for HS-SPME.
2.7. Headspace SPME procedure
For HS-SPME process, water samples (15.0 mL) were put in 25.0 mL glass vials containing a small magnetic bar and 3.0 g NaCl (20%, w/v). The needle of the SPME device containing the fiber was passed through the cap, and then the 3D-G coated fiber was pushed out and exposed to the headspace above the sample solution at 50 °C. After the extraction under stirring at 1000 rpm for 30 min, the fiber was withdrawn into the needle, removed from the vial and then, the needle was pierced to the GC injection port and the fiber was pushed out for thermal desorption at 250 °C for GC analysis. To eliminate the fiber carry-overs, the fiber was held in the injector for the whole run (17.3 min) before the next extraction. Prior to the first use each day, the fiber was first activated by keeping the fiber in the injection port at 250 °C for 30 min and then a blank analysis was made to verify that no interfering peaks exist in the fiber.2.8. Determination of enhancement factor
Enhancement factor (EF) was determined as the ratio of the chromatographic peak area of an analyte after SPME extraction to that before SPME (i.e. by direct injection of 1.0 μL of the standard solution with the analyte at the same concentration as that in the original water sample).12,33,343. Results and discussion
3.1. Characterization of the 3D-G-coated stainless steel fiber
The morphology of the 3D-G and the 3D-G-coated fiber was investigated by SEM (Fig. 2). The SEM images of the 3D-G at a low magnification (Fig. 2A) shows that the reduced GO sheets were well-assembled and interconnected to build a 3D network. The SEM images with high-magnifications (Fig. 2B and C) show the continuous and winkled structural characteristics of the 3D-G. Fig. 2D is a low-magnification image for the 3D-G coating, which shows that the coating possessed a homogeneous surface; the high magnification SEM image in Fig. 2E shows that the 3D-G coating had a porous structure. Such porous structure increased the available surface area of the fiber, which was beneficial for the extraction performance. | ||
| Fig. 2 Scanning electron micrographs of 3D-G and 3D-G-coated fiber. The 3D-G surface images at magnifications of (A) 1000×, (B) 20000×, and (C) 50000×. The surface images of the 3D-G-coated fiber at magnifications of (D) 400× and (E) 1000×. | ||
To evaluate the reduction level of GO and determine the composition of the prepared 3D-G coating, the XPS spectra for both GO and the 3D-G were measured as shown in Fig. 3. Based on XPS survey spectra, the percentages of oxygen in GO and 3D-G are around 36.82% and 16.43%, respectively (Fig. 3A and B). The decreased oxygen content in 3D-G indicated an efficient deoxygenation of GO and the formation of graphene.35 For the identification of the surface functionalizations, the deconvolutions of the C1s regions of the spectra for both GO and 3D-G were fitted by the four peaks as shown in Fig. 3C and D, which are assigned to C–C, C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–CO functional groups.12,35 The absorbance peaks of oxygen functionalities (C–O, CO, O–CO) in 3D-G sharply decreased after thermal process, indicating that most of the epoxide and hydroxyl functional groups on 3D-G were successfully removed due to the thermal reduction process.36 According to the FT-IR spectrum of GO in Fig. 4A, the most characteristic features for GO (Curve a) were the absorption bands corresponding to the CO of carbonyl stretching vibration at 1720 cm−1, the O–H deformation vibration at 1402 cm−1, the C–O stretching of epoxide group at 1071 cm−1 and the O–H broad peak at 3400 cm−1. As shown from the spectra of the GO and 3D-G in Fig. 4A, after thermal reduction, most of the oxygen functional groups of GO were removed in 3D-G (Curve b), which illustrated that GO was almost totally reduced by the thermal reduction.
O and O–CO functional groups.12,35 The absorbance peaks of oxygen functionalities (C–O, CO, O–CO) in 3D-G sharply decreased after thermal process, indicating that most of the epoxide and hydroxyl functional groups on 3D-G were successfully removed due to the thermal reduction process.36 According to the FT-IR spectrum of GO in Fig. 4A, the most characteristic features for GO (Curve a) were the absorption bands corresponding to the CO of carbonyl stretching vibration at 1720 cm−1, the O–H deformation vibration at 1402 cm−1, the C–O stretching of epoxide group at 1071 cm−1 and the O–H broad peak at 3400 cm−1. As shown from the spectra of the GO and 3D-G in Fig. 4A, after thermal reduction, most of the oxygen functional groups of GO were removed in 3D-G (Curve b), which illustrated that GO was almost totally reduced by the thermal reduction.
 | ||
| Fig. 3 Survey XPS spectra of (A) GO and (B) 3D-G; C1s XPS spectra of (C) GO and (D) 3D-G. | ||
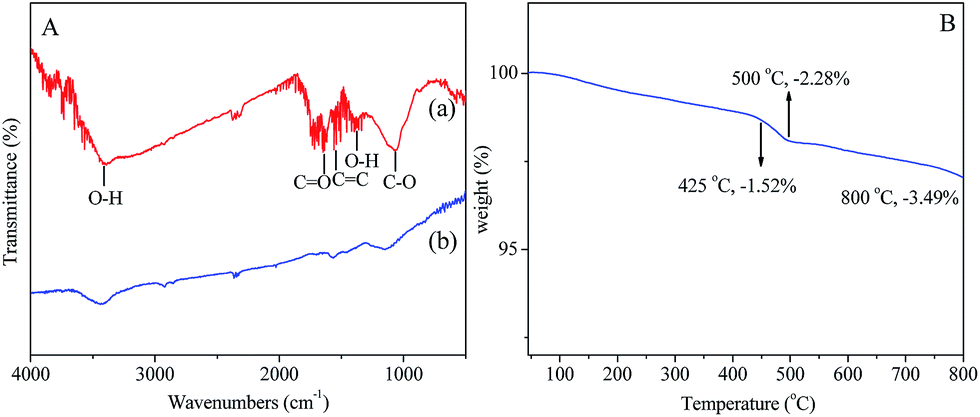 | ||
| Fig. 4 (A) FT-IR spectra of (a) GO, and (b) 3D-G; (B) thermogravimetric analysis (TGA) curve of 3D-G in nitrogen gas atmosphere; heating rate 10 °C min−1. | ||
The thermal resistance of the fiber coating is a very important parameter for SPME-GC applications. The thermal stability of the 3D-G coating was evaluated by the thermogravimetric analysis (TGA, see Fig. 4B). The samples were scanned within the temperature range from 25 to 800 °C at a rate of 10 °C min−1 under N2 atmosphere. It can be seen from Fig. 4B that 98.5% of the weight remained at 425 °C and 96.5% of the weight remained at 800 °C, indicating that the 3D-G can be thermally stable up to 425 °C and its thermal stability can endure the high temperature in the GC injector.
3.2. Optimization of the SPME procedures
Different experimental parameters that affect the extraction efficiency including extraction time and temperature, agitation speed, salt concentration, and desorption temperature and time, were respectively investigated and optimized in sequential order by using 15.0 mL of aqueous solution spiked with 0.1 μg L−1 each of the PAHs. | ||
| Fig. 5 Factors affecting the extraction efficiency for PAHs. (A) Effect of extraction temperature. (B) Effect of extraction time. (C) Effect of salt concentration. (D) Effect of agitation speed. | ||
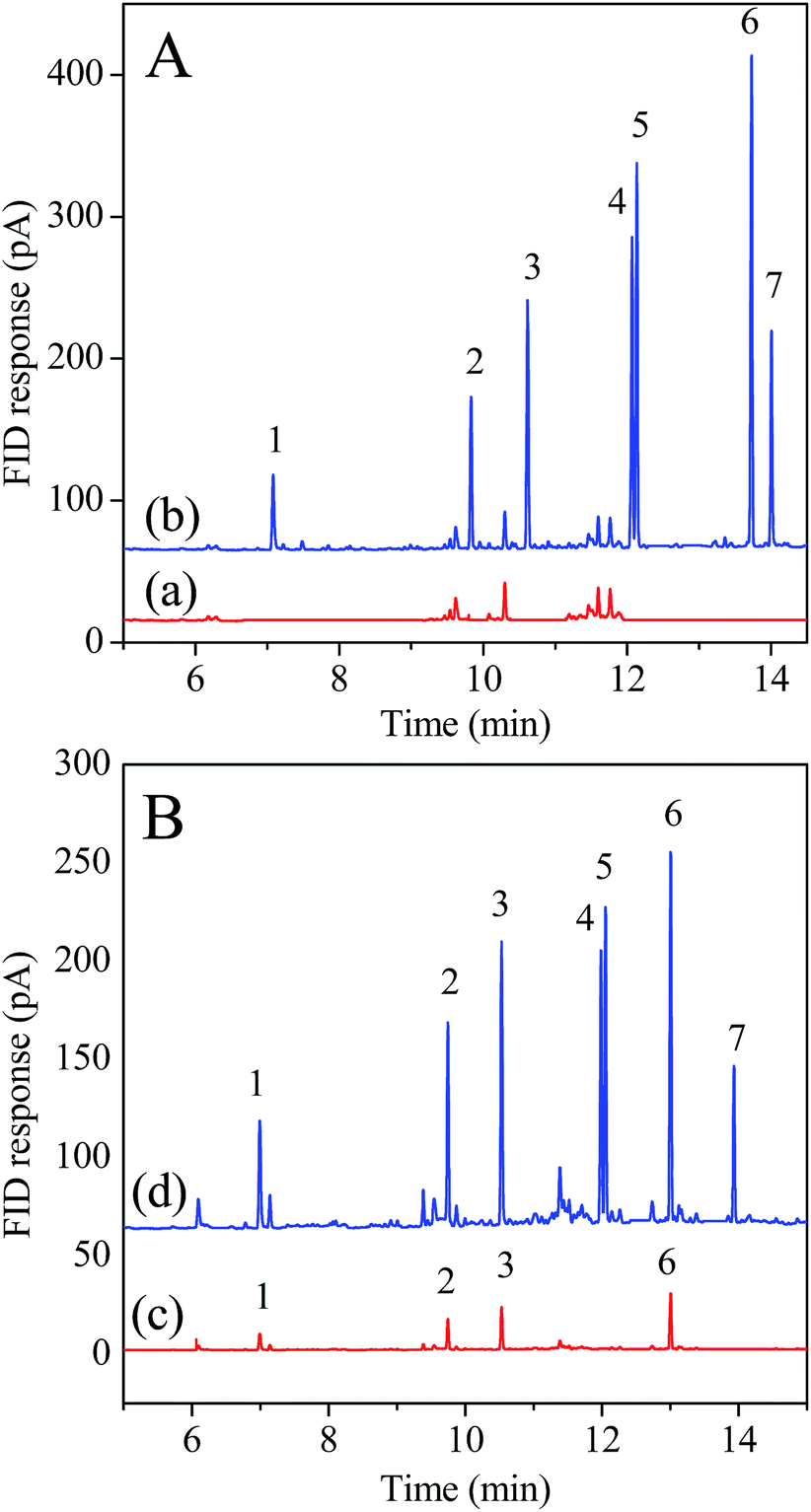 | ||
| Fig. 6 Chromatograms of extracts of PAHs from (A) pond water and (B) soil 2 samples obtained by the developed method. (a) Blank pond water sample and (b) spiked pond water sample at 100.0 ng L−1 level. (c) Blank farmland soil which close to a local thermal power plant (Soil 2) and (d) spiked Soil 2 sample at 50.0 ng g−1 level. Peak identifications: (1) NAP; (2) ANE; (3) FLU; (4) PHE; (5) ANT; (6) FLA; (7) PYR. | ||
Under the aforementioned optimized conditions, fiber lifetime, which influences the precision and accuracy of the SPME analysis, was also investigated by evaluating the extraction performance of the fiber after it was subjected to different extraction/desorption/conditioning cycles. As shown in Fig. S2B in the ESI,† for the same fiber, no obvious deteriorations in the extraction performance for the PAHs were found after 150 replicate extractions under the same optimized extraction conditions. These results suggest that the 3D-G fiber is quite stable and robust.
3.3. Possible adsorption mechanism of the 3D-G-coated fiber for analytes
To further understand the adsorption mechanism, three types of organic compounds with different physical chemical properties (such as hydrophobicity, electron polarizability, and polarity), namely PAHs, PAEs and n-alkanes, were tested. Some physical–chemical parameters of the PAHs, PAEs and n-alkanes are listed in Table 1. The adsorption affinity of the 3D-G coating to the tested compounds was evaluated by measuring their respective EFs for the HS-SPME and the results are also listed in Table 1. Generally, the EFs of the 3D-G coating to PAHs (EFs, 842-2458, Table 1) were much higher than those for the other compounds (EF ≤ 1500). The EFs increased with the number of the condensed rings of the PAHs (except for PYR) and were proportional to their respective hydrophobicity (KOW) and electron polarizability strength, implying the main role of the hydrophobic effect and π–π stacking interactions for the extraction. However, after a cross examination of the compounds from three different types, it can also be observed that a PAH with a close logKOW to a PAE or an n-alkane exhibited an extremely higher EF value than the other two. For example, FLA, DCHP and n-C11 have close logKOW values, but quite different EF values (2458, 1163 and 152) for them were obtained. The PAHs with logKOW values ranging from 3.36 to 5.31 and the PAEs (1.70 ≤ logKOW ≤ 5.64) were found to possess relatively higher EFs than the n-alkanes (6.10 ≤ logKOW ≤ 8.86). This observation revealed that π–π stacking interactions between the analyte and the 3D-G coating is more dominant than the hydrophobic interactions since 3D-G possesses highly delocalized conjugate system of π-electrons, which can form strong π–π stacking interactions with the aromatic rings in the PAHs or PAEs. The above results demonstrate that π–π stacking and hydrophobic interactions are the two main factors for the extraction of the 3D-G coating for the analytes.
| Analyte | Abbreviation | Structure | Molecular weight | Boiling point | logKOWa |
EFs |
|---|---|---|---|---|---|---|
| a K OW: n-octanol/water partition coefficients, taken from ref. 12 and 34. | ||||||
| Naphthalene | NAP |
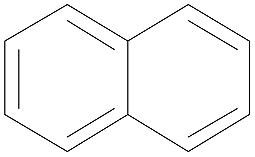
|
128 | 221 | 3.36 | 842 ± 13 |
| Acenaphthene | ANE |

|
154 | 279 | 3.73 | 969 ± 48 |
| Fluorene | FLU |
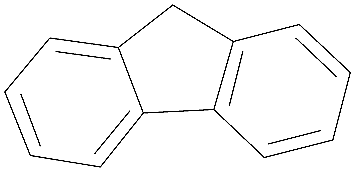
|
166 | 294 | 4.32 | 1330 ± 83 |
| Phenanthrene | PHE |

|
178 | 337 | 4.46–4.64 | 1793 ± 104 |
| Anthracene | ANT |

|
178 | 340 | 4.55 | 1837 ± 112 |
| Fluoranthene | FLA |

|
202 | 375 | 5.00 | 2458 ± 169 |
| Pyrene | PYR |

|
202 | 404 | 5.00 | 1904 ± 157 |
| Dimethyl phthalate | DMP |

|
194 | 283 | 1.70 | 251 ± 11 |
| Diethyl phthalate | DEP |
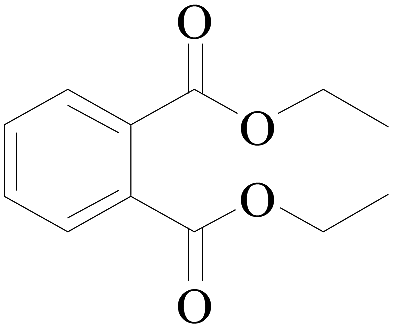
|
222 | 294 | 2.71 | 764 ± 36 |
| Dibutyl phthalate | DBP |

|
278 | 337 | 4.75 | 1163 ± 113 |
| Dicyclohexyl phthalate | DCHP |

|
330 | 426 | 5.64 | 1506 ± 120 |
| n-Undecane | n-C11 |

|
156 | 196 | 6.31 | 152 ± 7 |
| n-Dodecane | n-C12 |

|
170 | 216 | 6.83 | 202 ± 9 |
| n-Tridecane | n-C13 |

|
184 | 234 | 7.33 | 312 ± 18 |
| n-Tetradecane | n-C14 |

|
198 | 254 | 7.84 | 405 ± 24 |
| n-Pentadecane | n-C15 |

|
212 | 271 | 8.35 | 481 ± 29 |
| n-Hexadecane | n-C16 |

|
226 | 287 | 8.86 | 503 ± 26 |
3.4. Analytical figures of merit
Under the optimized conditions, the 3D-G coated fiber was evaluated for the extraction of seven PAHs, followed by the determination with GC-FID. For water samples, the establishment of the calibration curves for the analytes were performed in the concentration range of 10.0–5000.0 ng L−1 at nine different concentrations (10.0, 20.0, 50.0, 100.0, 200.0, 500.0, 1000.0, 2000.0, and 5000.0 ng L−1) spiked in PAHs-free water. For soil samples, the establishment of the calibration curves for the PAHs were performed in the concentration range of 1.0–200.0 ng g−1 at eight different concentrations (1.0, 2.0, 5.0, 10.0, 20.0, 50.0, 100.0 and 200.0 ng g−1) spiked in PAHs-free soil samples. The analytical characteristics of the developed method are presented in Table 2. For water sample, the linear response was observed in the concentration range from 10.0 to 1000.0 ng L−1 for ANT, FLA and PYR, from 20.0 to 5000.0 ng L−1 for FLU and PHE, and from 50 to 5000.0 ng L−1 for NAP and ANE, respectively, with r2 all larger than 0.9932. For soil sample, the linear response existed in the range from 1.0 to 100.0 ng g−1 for ANT and FLA, from 2.0 to 100.0 ng g−1 for PYR, from 2.0 to 200.0 ng g−1 for PHE and FLU, and from 5.0 to 200.0 ng g−1 for NAP and ANE, respectively, with r2 all larger than 0.9914. The limits of detection (LODs), calculated at a signal-to-noise ratio of 3, were in the interval between 2.0 and 10.0 ng L−1 for the water samples and 0.2–1.2 ng g−1 for soil samples, depending on compounds.| Analytes | Water samples | Soil samples | Reproducibility | |||||
|---|---|---|---|---|---|---|---|---|
| Linear range (ng L−1) | LODs (ng L−1) | r 2 | Linear range (ng g−1) | LODs (ng g−1) | r 2 | RSDa (%) | RSDb (%) | |
| a RSD, RSD of one fiber (n = 6). b RSD, RSD of fiber-to-fiber (n = 5). | ||||||||
| NAP | 50.0–5000.0 | 10.0 | 0.9969 | 5.0–200.0 | 1.2 | 0.9914 | 4.7 | 6.9 |
| ANE | 50.0–5000.0 | 9.0 | 0.9976 | 5.0–200.0 | 1.2 | 0.9959 | 5.0 | 8.3 |
| FLU | 20.0–5000.0 | 5.0 | 0.9983 | 2.0–200.0 | 0.5 | 0.9927 | 5.9 | 6.4 |
| PHE | 20.0–5000.0 | 4.0 | 0.9989 | 2.0–200.0 | 0.5 | 0.9980 | 4.6 | 8.2 |
| ANT | 10.0–1000.0 | 2.0 | 0.9963 | 1.0–100.0 | 0.2 | 0.9953 | 6.5 | 8.7 |
| FLA | 10.0–1000.0 | 2.0 | 0.9970 | 1.0–100.0 | 0.2 | 0.9929 | 7.1 | 9.4 |
| PYR | 10.0–1000.0 | 3.0 | 0.9932 | 2.0–100.0 | 0.5 | 0.9961 | 8.8 | 11.9 |
For the repeatability, the one same fiber was used for six replicate extractions of an aqueous sample containing 0.1 μg L−1 each of the PAHs under the same conditions, and the resulting relative standard deviations (RSDs) were below 8.8%. Five 3D-G coated fibers prepared in the same batch were used to evaluate the fiber-to-fiber reproducibility and the RSDs were lower than 11.9%. The 3D-G coated fibers gave lower LODs for PAHs than the literature reported commercial PDMS, PDMS/DVB and PA fibers (Table 3).
| Coating | Linear range (μg L−1) | LOD (ng L−1) | RSD (%) | Extraction time (min) | Analytical methods |
|---|---|---|---|---|---|
| 3D-G | 0.01–5.0 | 2.0–10.0 | 4.7–8.8 | 30 | GC-FID (present work) |
| PDMS (100 μm) | 0.1–50.0 | 10.0–120.0 | 3.7–27.0 | 60 | GC-MS39 |
| PDMS/DVB (65 μm) | 0.1–10.0 | 16.0–75.0 | 8.2–18.3 | 60 | GC-MS40 |
| PA (85 μm) | 0.1–10.0 | 12.0–138.0 | 5.7–9.3 | 60 | GC-MS40 |
| PDMS (7 μm) | 0.5–20 | 60.0–350.0 | 7.8–30 | 50 | GC-MS41 |
| Graphene | 0.05–200 | 4.0–50.0 | 2.8–9.4 | 50 | GC-FID34 |
| Multiwalled carbon nanotubes | 0.02–1000 | 9.0–13.0 | 1.7–3.6 | 15 | GC-FID42 |
| Polymeric ionic liquids | 0.5–20 | 50.0–250.0 | 9.2–29 | 50 | GC-MS41 |
3.5. Analysis of the PAHs in water and soil samples
The developed 3D-G coated fiber was applied to the SPME of PAHs in water samples and two farmland samples near a rubbish recycling center (Soil 1) and a local thermal power plant (Soil 2). As a result, no PAHs were detected in the local river and pond water samples. Then, the water samples were spiked with 100.0 ng L−1 each of the PAHs for the recovery measurements to evaluate the accuracy of the method. Good recoveries for the analytes were obtained with the current method, ranging from 80.1% to 102.6% (Table 4).| Analyte | Spiked (ng L−1) | River water (n = 3) | Pond water | Spiked (ng g−1) | Soil 1 | Soil 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Found (ng L−1) | Recovery (%) | RSD (%) | Found (ng L−1) | Recovery (%) | RSD (%) | Found (ng g−1) | Recovery (%) | RSD (%) | Found (ng g−1) | Recovery (%) | RSD (%) | |||
| a Not detected. | ||||||||||||||
| NAP | 0 | nda | nd | 0 | nd | 6.5 | ||||||||
| 100.0 | 82.9 | 82.9 | 6.5 | 80.1 | 80.1 | 13.0 | 50.0 | 39.4 | 78.8 | 6.7 | 52.1 | 91.2 | 8.0 | |
| ANE | 0 | nd | nd | 0 | 8.9 | 8.3 | ||||||||
| 100.0 | 89.6 | 89.6 | 6.7 | 89.4 | 89.4 | 9.2 | 50.0 | 54.2 | 90.6 | 7.6 | 53.4 | 90.2 | 5.7 | |
| FLU | 0 | nd | nd | 0 | nd | 11.7 | ||||||||
| 100.0 | 95.2 | 95.2 | 7.2 | 83.3 | 83.3 | 8.0 | 50.0 | 40.1 | 80.2 | 7.6 | 58.6 | 93.8 | 11.1 | |
| PHE | 0 | nd | nd | 0 | nd | nd | ||||||||
| 100.0 | 93.3 | 93.3 | 9.3 | 91.7 | 91.7 | 10.5 | 50.0 | 38.3 | 76.6 | 7.5 | 42.2 | 84.4 | 5.8 | |
| ANT | 0 | nd | nd | 0 | nd | nd | ||||||||
| 100.0 | 88.0 | 88.0 | 8.2 | 84.2 | 84.2 | 6.8 | 50.0 | 43.1 | 86.2 | 8.1 | 42.6 | 85.2 | 7.3 | |
| FLA | 0 | nd | nd | 0 | nd | 6.9 | ||||||||
| 100.0 | 95.7 | 95.7 | 9.0 | 102.6 | 102.6 | 8.7 | 50.0 | 43.6 | 87.2 | 9.5 | 53.3 | 92.8 | 10.0 | |
| PYR | 0 | nd | nd | 0 | nd | nd | ||||||||
| 100.0 | 89.4 | 89.4 | 11.3 | 89.0 | 89.0 | 8.9 | 50.0 | 46.4 | 92.8 | 3.9 | 40.9 | 81.8 | 7.0 | |
For soil samples, ANE was determined to be 8.9 ng g−1 in Soil 1. NAP, ANE, FLU and FLA were found in the range of 6.5–11.7 ng g−1 in Soil 2. The recoveries obtained at the spiking level of 50.0 ng g−1 each of the PAHs in soil ranged from 76.5% to 93.9%. The results show the feasibility of the 3D-G fibers for the SPME of trace PAHs in environmental samples. Fig. 6 shows the typical chromatograms for water and soil samples.
4. Conclusions
We have developed a simple strategy for the preparation of novel 3D-G coated SPME fibers based on sol–gel coating technique on a microstructured silver functionalized stainless steel wire. The 3D-G coated fiber exhibited excellent fiber-to-fiber repeatability and thermal stability, and it showed a good selectivity for aromatic analytes, especially for PAHs, via both π–π stacking and hydrophobic interactions. The 3D-G coated fiber was successfully used to extract trace PAHs in water and soil samples prior to GC-FID analysis. A wide linear range, low LODs and good recoveries for real samples indicated that the 3D-G coated fiber-based HS-SPME-GC-FID method was suitable for the determination of the analytes in environmental samples. Therefore, the method offers a sensitive and inexpensive alternative tool for the determination of trace PAHs in complicated samples.Acknowledgements
The authors acknowledge the financial support of the National Natural Science Foundation of China (no. 31171698, 31471643) and the Scientific Research Program of Hebei Education Department (QN2014133).References
- C. L. Arthur and J. Pawliszyn, Anal. Chem., 1990, 62, 2145–2148 CrossRef CAS.
- V. K. Ponnusamy and J. F. Jen, J. Chromatogr. A, 2011, 1218, 6861–6868 CrossRef CAS PubMed.
- J. Chen, J. Zou, J. Zeng, X. Song, J. Ji, Y. Wang, J. Ha and X. Chen, Anal. Chim. Acta, 2010, 678, 44–49 CrossRef CAS PubMed.
- F. Ghaemi, A. Amiri and R. Yunus, TrAC, Trends Anal. Chem., 2014, 59, 133–143 CrossRef CAS.
- Y. Li, M. Zhang, Y. Yang, X. Wang and X. Du, J. Chromatogr. A, 2014, 1358, 60–67 CrossRef CAS PubMed.
- J. Feng, H. Qiu, X. Liu, S. Jiang and J. Feng, TrAC, Trends Anal. Chem., 2013, 46, 44–58 CrossRef CAS.
- M. T. Jafari, M. Saraji and H. Sherafatmand, Anal. Chim. Acta, 2014, 814, 69–78 CrossRef CAS PubMed.
- D. Djozan, Y. Assadi and S. H. Haddadi, Anal. Chem., 2001, 73, 4054–4058 CrossRef CAS PubMed.
- J. Xu, J. Zheng, J. Tian, F. Zhu, F. Zeng, C. Su and G. Ouyang, TrAC, Trends Anal. Chem., 2013, 47, 68–83 CrossRef CAS.
- H. Bagheri, H. Piri-Moghadam and M. Naderi, TrAC, Trends Anal. Chem., 2012, 34, 126–139 CrossRef CAS.
- W. Zhang, Y. Sun, C. Wu, J. Xing and J. Li, Anal. Chem., 2009, 81, 2912–2920 CrossRef CAS PubMed.
- S. Zhang, Z. Du and G. Li, Anal. Chem., 2011, 83, 7531–7541 CrossRef CAS PubMed.
- J. W. Zewe, J. K. Steach and S. V. Olesik, Anal. Chem., 2010, 82, 5341–5348 CrossRef CAS PubMed.
- J. M. Jimenez-Soto, S. Cardenas and M. Valcarcel, J. Chromatogr. A, 2010, 1217, 3341–3347 CrossRef CAS PubMed.
- A. Rahimi, P. Hashemi, A. Badiei, P. Arab and A. R. Ghiasvand, Anal. Chim. Acta, 2011, 695, 58–62 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. Novoselov and A. Geim, Nat. Mater., 2007, 6, 183–191 CrossRef PubMed.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2009, 110, 132–145 CrossRef PubMed.
- M. Sun, J. Feng, Y. Bu, X. Wang, H. Duan and C. Luo, Talanta, 2015, 134, 200–205 CrossRef CAS PubMed.
- G. Chen, W. Weng, D. Wu, C. Wu, J. Lu, P. Wang and X. Chen, Carbon, 2004, 42, 753–759 CrossRef CAS.
- Y. B. Luo, B. F. Yuan, Q. W. Yu and Y. Q. Feng, J. Chromatogr. A, 2012, 1268, 9–15 CrossRef CAS PubMed.
- S. Zhang, Z. Du and G. Li, J. Chromatogr. A, 2012, 1260, 1–8 CrossRef CAS PubMed.
- H. Zhang and H. K. Lee, J. Chromatogr. A, 2011, 1218, 4509–4516 CrossRef CAS PubMed.
- J. Zou, X. Song, J. Ji, W. Xu, J. Chen, Y. Jiang, Y. Wang and X. Chen, J. Sep. Sci., 2011, 34, 2765–2772 CrossRef CAS PubMed.
- G. Zhang, X. Zang, Z. Li, Q. Chang, C. Wang and Z. Wang, Anal. Methods, 2014, 6, 2756 RSC.
- Y. Guo, S. Guo, J. Ren, Y. Zhai, S. Dong and E. Wang, ACS Nano, 2010, 4, 4001–4010 CrossRef CAS PubMed.
- S. Tong, Q. Liu, Y. Li, W. Zhou, Q. Jia and T. Duan, J. Chromatogr. A, 2012, 1253, 22–31 CrossRef CAS PubMed.
- Z. Niu, L. Liu, Y. Jiang and X. Chen, in Advanced Hierarchical Nanostructured Materials, ed. Q. Zhang and F. Wei, Wiley-VCH, Weinheim, 2014, pp. 291–350 Search PubMed.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- X. Jiang, Y. Ma, J. Li, Q. Fan and W. Huang, J. Phys. Chem. C, 2010, 114, 22462–22465 CAS.
- C. Wang, C. Feng, Y. Gao, X. Ma, Q. Wu and Z. Wang, Chem. Eng. J., 2011, 173, 92–97 CrossRef CAS.
- N. Chang, Z. Y. Gu, H. F. Wang and X. P. Yan, Anal. Chem., 2011, 83, 7094–7101 CrossRef CAS PubMed.
- M. Wu, L. Wang, B. Zeng and F. Zhao, J. Chromatogr. A, 2014, 1364, 45–52 CrossRef CAS PubMed.
- J. Fan, Z. Dong, M. Qi, R. Fu and L. Qu, J. Chromatogr. A, 2013, 1320, 27–32 CrossRef CAS PubMed.
- J. Tang, G. Chen, J. Yang, X. Zhou, L. Zhou and B. Huang, Nano Energy, 2014, 8, 62–70 CrossRef CAS.
- O. Akhavan, Carbon, 2010, 48, 509–519 CrossRef CAS.
- J. Ai and A. Chem, Anal. Chem., 1997, 69, 1230–1236 CrossRef CAS.
- W. Zhang, Y. Sun, C. Wu, J. Xing and J. Li, Anal. Chem., 2009, 81, 2912–2920 CrossRef CAS PubMed.
- E. Psillakis, A. Ntelekos, D. Mantzavinos, E. Nikolopoulos and N. Kalogerakis, J. Environ. Monit., 2003, 5, 135–140 RSC.
- D. Martin and J. Ruiz, Talanta, 2007, 71, 751–757 CrossRef CAS PubMed.
- L. Pang and J. F. Liu, J. Chromatogr. A, 2012, 1230, 8–14 CrossRef CAS PubMed.
- A. A. Matin, P. Biparva and M. Gheshlaghi, J. Chromatogr. A, 2014, 1374, 50–57 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The SEM of stainless steel wire and factors affecting the extraction efficiency for PAHs. See DOI: 10.1039/c5ra05616k |
| This journal is © The Royal Society of Chemistry 2015 |