
Assembly of 3-allyl-3-ethynyl-oxindole motifs via palladium(ii)-catalyzed quaternary allylation of 3-ethynyl-3-OBoc-oxindoles†
Abstract
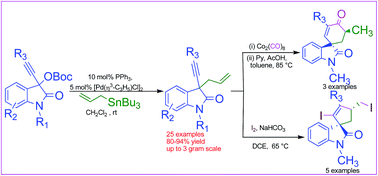
Palladium(II)-catalyzed C3-allylation of 3-ethynyl-3-OBoc oxindole derivatives was achieved for the first time to access highly functionalized 3-allyl-3-ethynyl substituted oxindole derivatives for a broad range of substrates in very good yields (up to 94%). The synthetic applications of the allylated products were explored by synthesizing five and six membered carbocyclic spirooxindoles in moderate to very good diastereoselectivities.
 Please wait while we load your content...
Please wait while we load your content...

