
Nanoporous silicon from low-cost natural clinoptilolite for lithium storage†
Rongrong Miaoa,
Jun Yang*a,
Yanan Wua,
Jiulin Wanga,
Yanna Nulia and
Wei Lub
aShanghai Electrochemical Energy Devices Research Center, School of Chemistry & Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: yangj723@sjtu.edu.cn; Fax: +86-21-54747667; Tel: +86-21-54747667
bDepartment of Chemical Engineering, University of Michigan, Ann Arbor, MI 48109, USA
First published on 22nd June 2015
Abstract
Despite the fact that silicon materials can be synthesized from various sources, deriving them from earth-abundant resources is of strategic significance for industrial processing. Here nanoporous silicon (pSi) was derived from earth-abundant natural clinoptilolite (NCLI) without complicated pretreatment. After surface carbon coating, the pSi–C composite displayed a superior stable capacity of ca. 1257 mA h g−1 and good cycling stability with 87.5% capacity retention on the 200th cycle versus the 3rd one, which benefit from its nanoporous structure, very small primary particle size of ∼10 nm and highly conductive carbon-matrix.
Introduction
Lithium ion batteries, as the most dominant power source currently, have been extensively used in portable devices, (hybrid) electric vehicles (HEV) and grid-scale stationary energy storage. However, the limitation in energy density and power density of existing lithium ion battery systems based on conventional graphite anodes and lithium metal oxide or phosphate cathodes (LiCoO2, LiMn2O4, LiFePO4) is becoming more and more prominent.1 During searching for new material with much higher energy density, silicon (Si) attracts tremendous attention due to its ten times higher theoretical capacity (4200 mA h g−1) than traditionally used graphite anodes.2 Whereas the main impediment of silicon as anode material is its severe particle pulverization and loss of electronic conductivity of the electrode originated from huge volumetric change during repeated lithiation/delithiation. Therefore, extensive research has been devoted to address the aforementioned issue. For instance, downsizing the dimensions of silicon to nanoscale could buffer the large (de)lithiation strains without fracture to some extent; other strategies are to design some inner free space in silicon to accommodate the large volume expansions and thereby well-designed silicon with various special structures have been proposed, including pomegranate-like hierarchical structure, lotus-root-like mesoporous structure, etc.3–5 In addition, carbon materials have also been put inside to form diverse Si–C composite, which is used to stabilize the whole structure and further enhance electronic conductivity of active material.6–8 Among these strategies, constructing porous silicon materials have been proven to be effective in improving cycle life and/or rate properties, but the high-cost silica source (e.g. SBA-15,9 KIT-6 (ref. 10)) and complicated synthesis process may hinder its large-scale production. As we all know, with the increase of transportation markets in hybrid electrical vehicles (HEVs), plug-in hybrid electrical vehicles (PHEVs) and electrical vehicles (EV), this broader and large-scale application raises issues of price and environment, as well as scalability, thus new designs of Si anodes with the desired combination of functional features have to be supplemented by using cost-effective silica precursor and preparation method, which is becoming extremely significant for its practical application.Recently, the developments on synthesizing porous or nano-scaled Si from low cost silica precursors look quite promising.11 Some synthetic silica precursors (e.g. modified stober silica or silica aerogels) are employed to synthesize porous silicon.12,13 Although excellent cyclability can be obtained, the preparation process of silica precursor seems to be complicated. Thereby, some natural silica precursors also attract attention. For instance, Wang et al.14 synthesized porous silicon by employing diatomaceous earth as cheap silica source. Nevertheless, the obtained porous silicon with carbon coating demonstrated relatively poor performance with a reversible capacity of 633 mA h g−1 after 30 cycles and silicon without carbon coating was even lower with only 376 mA h g−1. Other cheap silica sources such as sand or rice husk have also been selected and some silicon materials keeping the original silica structures were prepared successfully by magnesiothermic reduction process.15–17 However, the tedious pre-treatments of sand, such as milling, calcining and washing with HCl, HF and NaOH, may be unfavorable for its potential industrial application.17 Although silicon derived from rice husk exhibits high reversible capacity of 2790 mA h g−1 and long cycle life of 86% capacity retention over 300 cycles, the relatively low silica content (∼23% in mass) in rice husks largely reduces the single conversion yield of silicon.16 And the sophisticated pretreatments are also disadvantageous to large-scale production.
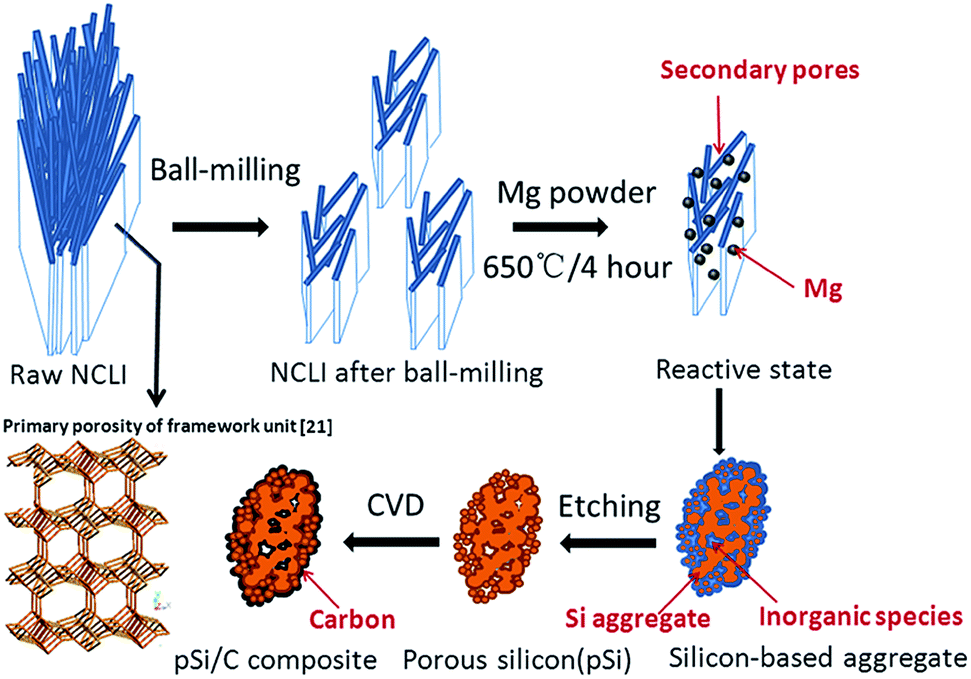
Herein, nanoporous silicon with primary particle size in ∼10 nm was directly synthesized from low-cost natural clinoptilolite (NCLI) through magnesiothermic reduction method with simple and facile pretreatment. Natural clinoptilolite is an earth-abundant resource of aluminosilicate available all over the world with extremely low-cost (RMB 2.0 kg−1). It is the most common natural zeolite and commercially used in wastewater processing, with cage-like structure containing AlO4 and SiO4 tetrahedra linked through the common oxygen atoms.18,19 It belongs to heulandite family with the molar Si/Al ratio above 4,20 and its specific crystal building is characterized by two channels running parallel to c-axis: a channel consisting of a 10-member ring with the size of 0.44–0.72 nm and a channel consisting of 8-member ring with the size of 0.41–0.47 nm, and a channel running parallel to a-axis consisting of an 8-member tetrahedral ring with the size of 0.40–0.55 nm (Scheme 1).21 These channels in NCLI framework structure form the primary micro-pores. Notably, the presence of secondary porosity is another interesting feature of NCLI, which is connected with cleavage of NCLI grains and other minerals in the NCLI rocks.22 This unique polymodal pore size distribution is favorable for the homogeneity of magnesiothermic reduction reaction and nanoporous silicon was successfully obtained even in the presence of alumina and other mineral species.
 | ||
| Scheme 1 Schematic illustration of the preparation of pSi–C composite. | ||
Compared with the reported silica templates for producing porous silicon, the silica source of natural clinoptilolite (NCLI) in this paper has several advantages: (i) the intricate cage and channel architectures in NCLI are beneficial to obtain porous structured silicon with nano-scaled primary particles in size of 10 nm, which accommodate the large volume expansion and ensure the facile strain relaxation; (ii) as-synthesized porous silicon after carbon modification exhibits excellent capacity retention and rate performance; (iii) NCLI inherits relatively high SiO2 content of 50–70 wt%, extremely low cost and abundant reserves in nature; (iv) there is no any complicated pre-treatment for the NCLI and the overall synthesis process is low-cost, energy-efficient and simple to be realized in large-scale production.
Experimental
Synthesis of porous silicon
Scheme 1 gives an overview of the synthesis procedure of the porous Si material from NCLI. Natural clinoptilolite (NCLI) containing ca. 68.6 wt% SiO2 from Zhejiang province, was firstly ball-milled with a Planetary Mono Mill P-6 (Fritsch, Germany) at a rotation speed of 500 rpm for 10 h to decrease the particle size and open the transfer channels (mass ratio of balls/material/water = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1).
:2:1).
The mixture of obtained NCLI powder and magnesium powder (Sinopharm Chemical Reagent Co. Ltd, 100–200 mesh) were loaded in an alundum boat with a molar ratio of Mg/SiO2 = 2.1. And then heated in a tube furnace at 650 °C for 4 h under Ar (95 vol%)/H2 (5 vol%) mixed atmosphere. The heating speed was kept at 2 °C min−1. The obtained brown powder was firstly immersed in 2 M HCl solution for 12 h to remove MgO and other impurities in NCLI. To further remove small amount of unreacted and surface-grown SiO2, 5 wt% HF/EtOH(10 vol%) solution was used and stirred for 15 min, then washed with distilled water and ethanol by filtration, finally vacuum-dried at 65 °C for 2 h.
Carbon was coated on the as-prepared porous silicon (pSi) by CVD method using toluene as the carbon source. The obtained pSi powder was loaded in an alundum boat and placed at the center of a quartz tube furnace. Next, the precursor gas (argon and toluene with the gas flow of 200 ml min−1) was introduced into the furnace for 30 min to flush away any oxygen in the reactor. Then, the furnace temperature was increased from room temperature to 800 °C at a rate of 10 °C min−1 and kept at 800 °C for 25 min. The furnace was cooled slowly to room temperature. At high temperature, the toluene decomposed quickly and carbon deposited onto the surface of pSi particles. The carbon content in the composite was controlled at 23.6 wt%.
Structure and morphology characterization
X-ray diffraction (XRD) patterns were recorded using Cu-Kα radiation at 40 kV with an X-ray Diffractometer (D/max-2200/PC, Rigaku). The morphologies of synthesized silicon and related composite materials were observed by a field emission scanning electron microscope (FESEM, JEOL JSM-7401F). Transmission electron microscope (TEM, JOEL JEM-100CX) was employed to characterize the microstructures of as-synthesized materials. The specific surface area and pore size distribution were determined from the results of N2 adsorption/desorption measurements using an ASAP 2020 Accelerated Surface Area and Porosimetry (Micromeritics Inc., USA). Inductively Coupled Plasma Mass Spectrometer (ICP, Agilent Inc. USA) analysis was conducted to determine the chemical component of obtained silicon. The carbon weight fractions in pSi–C samples were determined by evaluating the mass loss of pSi–C composites after calcination by thermogravimetric analysis (TGA, TA 2050). Raman spectrums were obtained by using a Thermo Scientific DXR Raman microscope with a laser wavelength of 532 nm at room temperature.Cells assembling and electrochemical tests
The electrochemical performances of the as-prepared silicon were tested using CR2016-type coin cells. The working electrodes were prepared by pasting a mixture of 60% active material, 20% Super P conductive carbon black (40 nm, Timical) and 20% styrene butadiene rubber/sodium carboxymethyl cellulose binder (SBR/SCMC, 3:5 by weight) onto pure Cu foil. The electrodes were cut to Φ12 mm sheets after being dried firstly, and then further dried at 60 °C in vacuum for 4 h. The weight of the electrode material was weighed by using XS 105DU analytical balances (METTLER TOLEDO, Inc.) and the readability is 0.01 mg. The CR2016 coin cells were assembled in an argon-filled glove box (MB-10 compact, MBraun) with 1 M LiPF6/EC + DMC (1:1 in volume ratio, ethylene carbonate (EC), dimethyl carbonate (DMC)) as electrolyte, including 10% fluoroethylene carbonate (FEC), ENTEK ET20-26 as separator, and pure lithium foil as counter electrode. The electrochemical performances were evaluated on a LAND battery test system (Wuhan Kingnuo Electronics Co., Ltd., China) at 25 °C with the cut-off voltage of 0.01 V versus Li/Li+ for discharge (Li insertion) and 1.2 V versus Li/Li+ for charge (Li extraction).
Result and discussion
Porous silicon material can be obtained directly from NCLI via magnesiothermic reduction process as shown in Scheme 1. The main impediments of magnesiothermic reduction process by using NCLI as precursor are the occurrence of side reaction and agglomeration of obtained products due to the intricate cage structure and small pore size distribution. During the magnesiothermic reaction, reducing agent Mg will become vapor state at elevated temperature and react with SiO2 in NCLI, the relative reactions are as follows,23| SiO2 (s) + 2Mg (g) → Si (s) + 2MgO (s) | (1) |
If Mg is in excess, it will react with product Si. In addition, unreacted SiO2 could further react with MgO as followed:
| Mg (g) + Si (s) → Mg2Si | (2) |
| SiO2 (s) + 2MgO (s) → Mg2SiO4 (s) | (3) |
MgO, Mg2Si and unreacted Mg can be removed easily by hydrochloric acid etching, while side reaction product Mg2SiO4 is hard to be removed. It is crucial to prevent the formation of the inert and insulating Mg2SiO4 and obtain the desired silicon with high quality.
NCLI is a natural micro- and mesoporous material with polymodal pore size distribution. The microporosity (primary porosity) is related to the clinoptilolite framework structure (as shown in Scheme 1), while the mesoporosity (secondary porosity) is mainly caused by cleavage phenomenon of the NCLI grains.22 In view of this polymodal porosity, it is a suitable silicon source for our work. However, pristine NCLI material without ball-milling process provides relatively small transport channels because of the close aggregation of thin-plates, which make it difficult for vapor Mg to penetrate into the inner part of particles uniformly. As a consequence, the side reaction with the product of Mg2SiO4 could take place, and the distribution of the particles is poor (black appearance revealed in Fig. S1(a)†). The capacity of pSi obtained in this case drops quickly and voltage polarization is serious (Fig. S2 and S3†). After high-energy mechanical milling (HEMM) pretreatment, the aggregating thin-plates are split off and the particle size decreases from ca. 20–50 μm to ca. 2 μm as shown in Fig. 2, which is favorable for the uniform penetration of Mg vapor into internal silicon oxide with a siginificantly shorter diffusion path. As a result, the side-reactions can be greatly suppressed and uniform Si powder with typical yellow appearance can be obtained (Fig. S1(b)†).
Apart from inhibiting side reaction, keeping a slow temperature ramp rate is very significant to obtain silicon with good performance as well. This finding is in accordance with Cui et al.16 In view of exothermic property of magnesiothermic reduction, local-heat accumulation may be incurred at high temperature ramp rate, which may further trigger agglomeration of obtained silicon and block its inner space. Therefore, after premixing with Mg powder, slow temperature ramp rate of 2 °C min−1 is necessary to ensure that the heat from the reaction can be dissipated sufficiently.
Silicon-based aggregation containing silicon and inorganic species (e.g. unreacted SiO2, MgO and other mineral species) was obtained after the magnesiothermic reduction process. Nano-scaled primary silicon particles can be formed from NCLI thin-plates, while silicon formed on sites of aggregated NCLI bars may go through further crystal growth into larger one or intergrowth into interconnected matrix. After being treated by HCl and HF solution to remove inorganic species, the porous structure with nano-scaled primary particles is constructed by residual silicon aggregation. At last a carbon layer was directly deposited on the surface to improve the electronic conductivity and promote solid electrolyte interface (SEI) formation for long-term cycles.
The X-ray diffraction (XRD) patterns in Fig. 1(a) compare the products obtained by magnesiothemic reduction of NCLI with and without ball-milling pretreatment. For both samples, peaks from Si and MgO are easily detected, indicating the successful magnesiothemic reduction of NCLI. On the other hand, the disappeared Mg2SiO4 peaks in the sample with ball-milling pretreatment indicates an effective inhibition of side reactions. Meanwhile, it's noted that the XRD peak intensity is decreased and full width data at half maximum (FWHM) intensity is broaden after ball-milling pretreatment, which reveals a weaker crystallinity and much smaller particle size according to Scherrer formula.24 The X-ray diffraction (XRD) patterns of acid-treated porous silicon obtained from NCLI with pretreatment are shown in Fig. 1(b). The peaks at 28°, 47°, 56°, 69°and 76°can be readily indexed as the (111), (220), (311), (400), and (331) planes of Si crystals, respectively (JCPDS card 27-1402), suggesting that the target product can be obtained through choosing appropriate ratio of reactive materials in magnesiothermic reduction reaction and impurities such as magnesia, residual SiO2, magnesium and other mineral species can be removed by acid etching process. The Scherrer analysis based on silicon patterns in Fig. 1(b) reveals a crystallite size of 12.3 nm, which is consistent with an estimate from TEM. Moreover, because silicon is directly formed from NCLI, the purity is a subject which can't be ignored. According to ICP result, the main impurity in obtained Si is determined to be Al (∼3 wt%), which comes from the cage structure of NCLI. Other impurities (K, Na, Fe, Ca and Mg) are common to mineral substance and their total concentration is less than 1%. It appears that these impurities do not exert an obvious negative effect.
 | ||
| Fig. 1 XRD patterns of (a) products obtained by magnesiothermic reduction from NCLI with and without ball-milling pretreatment (before acid etching); (b) acid-treated porous silicon obtained from NCLI with pretreatment. | ||
SEM and TEM characterizations are used to investigate the morphology and structure of the samples. Fig. 2 shows morphologies of NCLI with/without ball-milling pretreatment and the obtained silicon samples. It can be seen from Fig. 2(a) that pristine NCLI contains particles with a very broad size distribution from less than 5 μm to ∼50 μm. From Fig. 2(b) we can see the lamellar texture of NCLI with overlapping plates or bars. The thin-plates are of 30–50 nm in thickness and 700–800 nm in length. Furthermore, the transport channels of NCLI (represented as mesopores) caused by splitting of these face-to-face arranging thin plates can be found obviously. However, the size of these mesopores is relatively small due to the close aggregation of planes or bars. After ball-milling, the particles of NCLI were broken down to smaller ones with a homogeneous particle distribution around 2 μm in size (Fig. 2(c)). Moreover, the cleavage degrees are aggrandized significantly as a result of mechanical powdering process and transport channels are opened up (as shown in Fig. 2(d)), which is in favour of the uniform penetration of vapour Mg. As shown in Fig. 2(e) and (f), the sponge-like silicon composed of highly porous network of interconnected crystalline silicon is finally obtained after magnesiothemic reduction and acid etching.
 | ||
| Fig. 2 SEM images of (a and b) NCLI precursor; (c and d) NCLI after ball-milling; (e and f) as-synthesized silicon after acid etching. | ||
The porous properties of NCLI with ball-milling treatment and the obtained pSi are further examined by Brunauer–Emmett–Teller (BET) measurement (Fig. 3). As exhibited by Fig. 3(a), polymodel pores around 4 nm and 17 nm are observable for NCLI with ball-milling. The broad pore size distribution (inset in Fig. 3(a)) may be associated with the different cleavage degree between thin plates in NCLI grains. The nitrogen adsorption–desorption result shown in Fig. 3(b) indicates the presence of pores in the obtained Si. Specific surface area of the pSi is 148.04 m2 g−1, which is much higher than that of the as-received NCLI (29.5 m2 g−1) and NCLI after ball-milling (32.1 m2 g−1). And the curves at a relative pressure of 0.7–0.9 can be classified as a type IV isotherm characteristic of mesoporous materials.25 Furthermore, the BJH pore diameter distribution (inset in Fig. 3(b)) reveals that the main pore distribution lies in ∼10 nm, with a cumulative pore volume of 0.692 cm g−1. The pores arise partly from the nature NCLI structure and partly from Al and MgO completely removed via chemical etching. After carbon coating, the main pore distribution decreases from ∼10 nm to ∼8 nm due to the carbon deposition within the nano-pores via the penetration of toluene molecules, as determined by BJH pore diameter distribution in Fig. S4.† The broad range of pore size is attributable to the random pore distribution in NCLI. They could effectively accommodate the volume changes of silicon during charge and discharge.
 | ||
| Fig. 3 Nitrogen adsorption–desorption isotherm linear plot of (a) NCLI-after ball-milling; (b) obtained porous silicon and the insets are Barrett–Joyner–Halenda (BJH) pore size distribution. | ||
The porous structure of as-prepared silicon can be further observed by TEM images in Fig. 4. As shown in Fig. 4(a), nanopores with a broad range of 3–20 nm are formed. The enlarged image of selected area in Fig. 4(a) confirms that the nanoporous structure is formed by primary silicon crystallites with uniform size distribution of ∼10 nm, which is in good agreement with the crystalline size based on the Scherrer analysis of Fig. 1(b). The high internal porosity and small size of primary silicon particle can better accommodate the volume change of Si and improve its stability. The lattice fringes with the interplanar spacing of 3.1 Å correspond to Si (111) plane (inset in Fig. 4(b)), which further prove that the silicon was successfully obtained from NCLI. EDS spectrum analysis (Fig. 4(c)) demonstrated the presence of Al, which is in agreement with the results of ICP. Because Al exists in the frame structure of NCLI, it may be imbedded in synthesized Si so that HCl can't access it during etching, which finally results in trace amount of residual Al.
 | ||
| Fig. 4 (a)TEM image of pSi; (b) highlighted image of the selected areas in (a) and inset is HR-TEM image of obtained silicon; (c) EDS spectrum captured for the region shown in (a); (d) TEM image of pSi–C composite. | ||
Based on the poor-conducting property of silicon, a surface modification method of CVD was employed to further enhance the electrochemical behavior of pSi. Toluene was used as carbon source to acquire the carbon coated nanoporous silicon composite (pSi–C) with the carbon content of 23.6 wt%. The resultant pSi–C composite was investigated by Raman spectroscopy. The two peaks at 508 cm−1 and 840 cm−1 in Fig. 5(a) are associated with crystalline Si. The other two additional peaks at 1333 cm−1 and 1589 cm−1 are attributed to the D (disordered) and G (graphite) band of carbon, respectively, indicating the presence of carbon. The ratio of D band to G band is estimated to be ∼0.92, demonstrating a relatively low graphitization degree for the pSi–C composite. Fig. 5(b) presents the morphology of pSi–C composite. The particles became larger as a result of carbon deposition compared with the primary silicon crystallites with the size of ∼10 nm. The carbon layer can be further studied through TEM and a fairly homogeneous carbon layer with a thickness of ca. 5 nm is strongly bound to the surface of the pSi–C composite (Fig. 4(d)). We can also see the lattice fringes clearly with interplanar spacing of 3.1 Å, corresponding to Si (111) plane.
 | ||
| Fig. 5 (a) Raman spectrum of pSi–C composite; (b) SEM images of pSi–C composite. | ||
Fig. 6 shows the voltage profiles of the pSi and pSi–C composite cycled between 0.01 and 1.2 V at 0.05C for the first two cycles and 0.2C for the following cycles. The first discharge (lithiation) curve displays a long flat plateau below 0.1 V, which corresponds to the Li-alloying process of crystalline Si to form amorphous LixSi phase.26 Afterwards, the discharge and charge curves show the characteristic of amorphous Si. pSi electrode delivers the first charge and discharge capacities of ca. 1768.5 mA h g−1 and ca. 2884.4 mA h g−1 respectively (shown in Fig. 6(a)), corresponding to its columbic efficiency of 61.3%. The relatively low columbic efficiency may be attributed to mass defected points and abundant surface area of pSi, which consumed more charges to form the solid electrolyte interphase (SEI) layer during the first discharge process. During cycling, the voltage polarization of pSi becomes apparent and its specific capacity drops gradually. To enhance the electric conductivity of silicon and stabilize the whole structure, carbon layer was coated on the surface of pSi. As shown in Fig. 6(b), the first charge and discharge capacities of pSi–C composite electrode are ca. 1302.5 mA h g−1 and ca. 1835.7 mA h g−1 respectively, corresponding to its columbic efficiency of 70.9%. The specific charge–discharge capacities were calculated according to the whole weight of silicon and carbon for pSi–C composite electrode. After carbon coating, some nanopores in pSi will be filled with carbon and the surface area was decreased, which may associate with the slight improvement of columbic efficiency for pSi–C composite electrode. In addition, the reproducible shape of curves for pSi–C composite electrode after the initial Li intercalation and de-intercalation process indicates the high electrochemical reversibility.
 | ||
| Fig. 6 Charge–discharge curves of the (a) pSi and (b) pSi–C composite for the 1st, 2nd, 20th and 50th cycles at 0.05C for the initial two cycles and at 0.2C for the following cycles (the specific charge–discharge capacities were calculated according to the whole weight of silicon and carbon for pSi–C composite electrode). | ||
To further characterize the electrochemical properties of pSi–C composite electrodes, cyclic voltammetry (CV) was measured at a scan rate of 0.1 mV s−1 over the potential window of 0–1.5 V versus Li/Li+(shown in Fig. 7). In the first cathodic scan, a broad peak at 1.26 V is ascribed to the formation of a SEI layer at the pSi–C/electrolyte interface, which disappears from the subsequent cycles. The distinct current peak below 0.13 V corresponds to Li insertion into crystalline Si to form amorphous LixSi phase.27,28 A new cathodic peak at ∼0.2 V appears from the second cycle, which could be related to alloying reaction in the activated electrode with amorphous Si phase. In the anodic scan, the two peaks at 0.32 V and 0.47 V are observed in the first cycle and become more distinct in the following ones. These two peaks correspond to delithiation of amorphous LixSi to α-Si.29 Notably, after six CV cycles, the profiles became almost overlapped, which further verifies the good cycling stability of pSi–C electrode.
 | ||
| Fig. 7 Cyclic voltammograms of the pSi–C composite electrode measured in the voltage region of 0–1.5 V with a scan rate of 0.1 mV s−1. | ||
Fig. 8(a) compares the reversible capacity and stability for the pSi obtained from NCLI, pSi–C composite and commercial nano-silicon (50–200 nm). Although the nano-Si electrode exhibits the highest initial capacity near 2300 mA h g−1, a rapid capacity fade occurs from the very start cycling due to the particle pulverization and electric disconnection of the material. The pSi electrode presents the initial capacity of ca. 1768.5 mA h g−1 and its capacity retention at the 200th cycle is ∼65.4% against at the 3rd cycle. In contrast, pSi–C composite delivers superior electrochemical reversibility and its capacity of 87.5% can be retained under the same condition. The typical coulombic efficiency of pSi–C composite approaches 99.4%. Its good cycling stability can be ascribed to the rich porous structure, nano-scaled primary particle size and conducting net-work formed by carbon layer, which buffer the huge volume change of silicon effectively and improve the mechanical integrity and conductivity of composite material. Meanwhile, this pSi–C composite electrode shows excellent rate performance. Fig. 8(b) shows that the capacity reposefully declines from ca. 1457.7 to ca. 907.7 mA h g−1 as the C-rate increased in stages from 0.05C to 2C. When the C-rate turns back to 0.05C, the capacity can recover to ca. 1432.7 mA h g−1. However, the capacity of the pSi electrode drastically declines from ca. 1854.1 mA h g−1 to ca. 672.9 mA h g−1 with 63.7% capacity loss under the same conditions.
 | ||
| Fig. 8 (a) Cycling performance of pSi, pSi–C composite and commercial nano-silicon electrodes at 0.05C for the initial two cycles and at 0.2C for the following cycles; (b) reversible capacities of pSi and pSi–C composite electrodes cycled at different C-rates from 0.05C to 2C (the specific charge–discharge capacities were calculated according to the whole weight of silicon and carbon for pSi–C composite electrode). | ||
The electrochemical impedance spectroscopy (EIS) of the pSi and pSi–C composite electrode was investigated to gain further insights into the improved cycling performance (Fig. 9). The depressed semicircle in the high-middle frequency region is assigned to the overlap between the SEI film and the interfacial charge transfer impedance, while the oblique straight line in the low frequency region corresponds to the ion diffusion within the anodes. The Nyquist plots of the two electrodes reveals that the total SEI and charge transfer resistances decrease from the first cycle to the 60th cycle, which may be attributed to the enhanced electronic conductivity due to trapped lithium in the electrode.30 Moreover, the pSi–C composite electrode exhibits apparent smaller high frequency semicircle and become basically stable after 60 cycles, indicating that pSi–C composite is more favorable for establishing a stable electronic and ionic transport pathways at the electrode–electrolyte interface. The EIS results can explain why pSi–C composite electrode possesses better capacity retention and rate performance than pSi electrode.
 | ||
| Fig. 9 Impedance plots for the pSi and pSi–C composite electrode at 1.2 V vs. Li/Li+ after different cycles. | ||
Conclusions
In summary, nanoporous silicon material with a sponge-like morphology was successfully prepared from low-cost earth-abundant natural clinoptilolite (NCLI). The polyporous nature of NCLI is beneficial to its reduction to silicon with porous structure. The nanopores serve as buffer zone to effectively accommodate the volume variations of silicon during lithiation and the well-dispersed primary particles in size of ∼10 nm ensure the facile strain relaxation. After surface carbon coating, the pSi–C composite displays a superior stable capacity of ca. 1257 mA h g−1 and good stability with 87.5% capacity retention on the 200th cycle versus the 3rd one (compared at the same current rate). The improvements can be ascribed to the nano-pores, very small primary particle size in ∼10 nm and highly conductive carbon layer. The simple and facile synthesis technique, low cost silica precursor and excellent electrochemical performance make this Si composite a promising candidate for large-scale production of the next-generation high capacity lithium-ion battery anode material.Acknowledgements
This work was supported by National Key 973 Program of the PRC (no. 2014CB932303) and SJTU-UM Joint Research Project.Notes and references
- H. Wu and Y. Cui, Nano Today, 2012, 7, 414–429 CrossRef CAS PubMed.
- H. K. Liu, Z. Guo, J. Wang and K. Konstantinov, J. Mater. Chem., 2010, 20, 10055–10057 RSC.
- Y. G. Guo, J. S. Hu and L. J. Wan, Adv. Mater., 2008, 20, 2878–2887 CrossRef CAS PubMed.
- J. Cho, J. Mater. Chem., 2010, 20, 4009–4014 RSC.
- N. Liu, Z. Lu, J. Zhao, M. T. McDowell, H.-W. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed.
- S.-H. Ng, J. Wang, D. Wexler, K. Konstantinov, Z.-P. Guo and H.-K. Liu, Angew. Chem., Int. Ed., 2006, 45, 6896–6899 CrossRef CAS PubMed.
- Y. Zhao, X. Liu, H. Li, T. Zhai and H. Zhou, Chem. Commun., 2012, 48, 5079–5081 RSC.
- R. C. de Guzman, J. Yang, M. M.-C. Cheng, S. O. Salley and K. Y. Simon Ng, J. Power Sources, 2014, 246, 335–345 CrossRef CAS PubMed.
- H. Jia, P. Gao, J. Yang, J. Wang, Y. Nuli and Z. Yang, Adv. Energy Mater., 2011, 1, 1036–1039 CrossRef CAS PubMed.
- P. Gao, H. Jia, J. Yang, Y. Nuli, J. Wang and J. Chen, Phys. Chem. Chem. Phys., 2011, 13, 20108–20111 RSC.
- M. Ge, Y. Lu, P. Ercius, J. Rong, X. Fang, M. Mecklenburg and C. Zhou, Nano Lett., 2014, 14, 261–268 CrossRef CAS PubMed.
- W. Wang, Z. Favors, R. Ionescu, R. Ye, H. H. Bay, M. Ozkan and C. S. Ozkan, Sci. Rep., 2015, 5, 1–5 Search PubMed.
- Q. Li, L. Yin and X. Gao, RSC Adv., 2015, 5, 35598–35607 RSC.
- L. Shen, X. Guo, X. Fang, Z. Wang and L. Chen, J. Power Sources, 2012, 213, 229–232 CrossRef CAS PubMed.
- N. Hai, I. Grigoriants and A. Gedanken, J. Phys. Chem. C, 2009, 113, 10521–10526 CAS.
- N. Liu, K. Huo, M. T. McDowell, J. Zhao and Y. Cui, Sci. Rep., 2013, 3, 1–7 Search PubMed.
- Z. Favors, W. Wang, H. H. Bay, Z. Mutlu, K. Ahmed, C. Liu, M. Ozkan and C. S. Ozkan, Sci. Rep., 2014, 4, 1–7 Search PubMed.
- K. Pavelić, M. Hadžija, L. Bedrica, J. Pavelić, I. Đikić, M. Katić, M. Kralj, M. H. Bosnar, S. Kapitanović and M. Poljak-Blaži, J. Mol. Med., 2001, 78, 708–720 CrossRef.
- Y. F. Wang, F. Lin and W. Q. Pang, J. Hazard. Mater., 2007, 142, 160–164 CrossRef CAS PubMed.
- Y. F. Tao, Y. Qiu, S. Y. Fang, Z. Y. Liu, Y. Wang and J. H. Zhu, J. Hazard. Mater., 2010, 180, 282–288 CrossRef CAS PubMed.
- M. W. Ackley, R. F. Giese and R. T. Yang, Zeolites, 1992, 12, 780–788 CrossRef CAS.
- M. Sprynskyy, R. Golembiewski, G. Trykowski and B. Buszewski, J. Phys. Chem. Solids, 2010, 71, 1269–1277 CrossRef CAS PubMed.
- W. Chen, Z. Fan, A. Dhanabalan, C. Chen and C. Wang, J. Electrochem. Soc., 2011, 158, A1055–A1059 CrossRef CAS PubMed.
- H. Borchert, E. V. Shevchenko, A. Robert, I. Mekis, A. Kornowski, G. Grübel and H. Weller, Langmuir, 2005, 21, 1931–1936 CrossRef CAS PubMed.
- S. Brunauer, L. S. Deming, W. E. Deming and E. Teller, J. Am. Chem. Soc., 1940, 62, 1723–1732 CrossRef CAS.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- M. Obrovac and L. Krause, J. Electrochem. Soc., 2007, 154, A103–A108 CrossRef CAS PubMed.
- X. Feng, J. Yang, P. Gao, J. Wang and Y. Nuli, RSC Adv., 2012, 2, 5701–5706 RSC.
- L. Zhang, W. Hao, H. Wang, L. Zhang, X. Feng, Y. Zhang, W. Chen, H. Pang and H. Zheng, J. Mater. Chem. A, 2013, 1, 7601–7611 CAS.
- X. Chen, K. Gerasopoulos, J. Guo, A. Brown, C. Wang, R. Ghodssi and J. N. Culver, Adv. Funct. Mater., 2011, 21, 380–387 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra08622a |
| This journal is © The Royal Society of Chemistry 2015 |