
Retracted Article: Design and synthesis of VEGFR-2 tyrosine kinase inhibitors as potential anticancer agents by virtual based screening†
Abstract
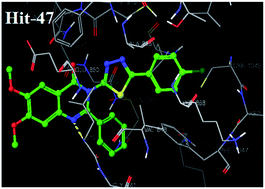
Vascular endothelial growth factor receptor-2 (VEGFR-2) plays a crucial role in cancer angiogenesis. A library of 6,7-dimethoxy quinazoline was prepared using a ligand based drug design approach and passed through different filters of virtual screening such as a docking study and Lipinski's rule. Twenty virtually screened compounds were synthesized and investigated against VEGFR-2 kinase and human umbilical vein endothelial cells (HUVEC) in vitro. Virtually screened compound 47 having 4-chlorophenyl-1,3,4-thiadiazole substitution at 3rd position of 6,7-dimethoxy-2-phenylquinazolin-4-(3H)-one exhibited the most promising activity, with IC50 values of 3.8 nm and 5.5 nm against VEGFR-2 tyrosine kinase and the HUVEC cell line. Docking simulation supported the initial pharmacophoric hypothesis and suggested a common mode of interaction at the ATP-binding site of VEGFR-2 demonstrating that compound 47 is a potential agent for cancer therapy that deserves further research.
 Please wait while we load your content...
Please wait while we load your content...

