
Combination of NH2OH·HCl and NaIO4: an effective reagent for molecular iodine-free regioselective 1,2-difunctionalization of olefins and easy access of terminal acetals†
Nirnita Chakrabortya,
Sougata Santrab,
Shrishnu Kumar Kunduc,
Alakananda Hajraa,
Grigory V. Zyryanovbd and
Adinath Majee*a
aDepartment of Chemistry, Visva-Bharati (A Central University), Santiniketan 731235, India. E-mail: adinath.majee@visva-bharati.ac.in; Fax: +91 3463 261526; Tel: +91 3463 261526
bDepartment of Organic Chemistry, Chemical Technological Institute, Ural Federal University, 19 Mira Str., 620002 Yekaterinburg, Russian Federation
cDepartment of Chemistry, Jhargram Raj College, Midnapore (W), Jhargram-721507, India
dI. Ya. Postovskiy Institute of Organic Synthesis, Ural Division of the Russian Academy of Sciences, 22 S. Kovalevskoy Str., 620219 Yekaterinburg, Russian Federation
First published on 23rd June 2015
Abstract
We have demonstrated a new application of our oxidizing reagent, a combination of NH2OH·HCl and NaIO4, in the first generalized regioselective 1,2-difunctionalization of olefins. It is a general method for the preparation of β-iodo-β′-hydroxy ethers, β-iodo ethers, β-iodohydrin, and β-iodo acetoxy compounds using different reaction media. The reactions are highly regioselective, always affording Markovnikov's type addition products. The methodology is also applicable for the easy access of terminal acetals. Molecular iodine-free synthesis, room temperature reaction conditions, high yields, use of less expensive reagents, mild reaction conditions, broad applicability of nucleophiles, and applicability for gram-scale synthesis are the notable advantages of this present protocol.
Introduction
The reaction of alkenes mainly difunctionalization is a fine approach adopted in organic synthesis. It has been widely studied and utilized in various techniques for functional group interconversions.1 Among them the halohydrin, β-iodo ether and β-iodo acetoxy compounds play a crucial role in the fields of drug scaffolds, synthetic organic chemistry, medicinal and industrial chemistry as well as material sciences.2 They are also the key intermediates in the synthesis of several halogenated marine natural products.2 The vicinal dihaloalkanes are formed by the electrophilic halogenation of alkenes.3 When the halogenation of the alkene is carried out in a nucleophilic solvent such as water, alcohols, carboxylic acids, nitriles, etc., difunctionalized products like halohydrins, β-haloethers, β-haloesters, etc. are obtained. This process is known as ‘cohalogenation’ and this is very important strategy to provide useful products for diverse synthetic applications.4The formation of halohydrins from alkenes is a well-established method.5 On the other hand, halohydrins are also useful intermediates in epoxide synthesis in both laboratory and industrial scales.6 The formation of chlorohydrins and bromohydrins by the reaction of alkenes and dilute aqueous solutions of the halogens undergoes smoothly7 but the formation of iodohydrins is not so smooth because of the reversibility of the addition of iodine to the double bond. An iodide ion scavenger such as AgNO3, HgO,8 CuO·HBF4 (ref. 9) or an oxidizing agent10 is essential for the formation of iodohydrins. The ring-opening of epoxides by hydrogen halides is also the most common procedure for the preparation of halohydrins. Hydrogen halides and hypohalite–water are the conventional reagents for epoxide ring opening to halohydrins.11 The main disadvantage of the methodologies for synthesis of halohydrins is to synthesize the epoxide first by employing the traditional methodologies like the reaction of alkenes with peracids/bases,12 O2 or H2O2 using a metal-based catalytic system13 or zeolites,14 H2O2/auxiliaries (nitriles, carbodiimides, etc.).15 Few methodologies have also been reported for the preparation of halohydrin compounds using mild reaction conditions with limited nucleophiles.16
β-Iodo ethers are important intermediates for stereoselective radical reactions17 as well as synthesis of E- or Z-alkenes with good to moderate diastereoselectivity.18 A number of methodologies for the synthesis of this important framework has been developed by various groups. Among these the most important approaches are the reactions of alkenes with I(py)2BF4,19 excess amount of iodine,20 diacetoxyiodine(I) complexes,21 I2/clays,22 I2/ultrasound,23 N-haloimides,24 N-halosaccharin,25 N-iodosuccinimide/alcohols,26 triiodoisocyanuric acid27 and IBX-I2.28
Regardless of their efficiency and reliability, most of these methods suffer from one or more of these disadvantages such as using expensive reagents and catalysts, long reaction times, requirement of inert atmosphere, harsh reaction conditions and mostly use of molecular iodine as iodo source. Although molecular iodine is a versatile reagent in organic synthesis; it is highly corrosive, toxic, and sublimable, making its use somewhat unattractive.29 Again, it is important to note that all these methods8–28 are not general for the preparation of halohydrin, β-iodo ether and β-iodo acetoxy compounds using the same reaction conditions; varying the nucleophilic medium like water, alcohol, carboxylic acid etc. respectively. Therefore, finding a general and efficient methodology for the synthesis of iodohydrins and β-iodo ethers in terms of using basic chemicals as starting materials, increasing efficiency, operational simplicity, mild reaction conditions, and economic practicability is highly desirable.
In continuation of our research in organic synthesis30 herein, we report a mild and efficient approach for the regioselective synthesis of various β-iodo-β′-hydroxy ethers, β-iodo ethers, iodohydrins, and β-iodo acetoxy compounds from alkenes using the combination of NaIO4 and NH2OH·HCl at room temperature within a short reaction time (Scheme 1b). Recently we have reported a mild and efficient approach for oxidation of alcohols to corresponding carbonyl compounds (Scheme 1a).31 Based on this report we can suggest that the in situ generated iodine undergoes the addition to the double bond to form iodonium ion which in presence of nucleophilic solvents like alcohols, water, carboxylic acids etc. might afford the corresponding β-iodo-β′-hydroxy ether, β-iodo ethers, iodohydrins, and β-iodo acetoxy compounds.
 | ||
| Scheme 1 Reaction of styrene with ethylene glycol to synthesize β-iodo-β′-hydroxy ether. | ||
Results and discussion
During our initial study, readily available styrene 1a was taken as a model substrate using NaIO4 (2 equiv.) and NH2OH·HCl (4 equiv.) as reagent in ethylene glycol solvent. The reaction proceeded smoothly at room temperature and the product 2-(2-iodo-1-phenylethoxy)ethanol (2a) was isolated in 86% yield within 30 min. Encouraged by this result our attention was focused on the optimization of the reagents ratios. First of all, we used 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 proportion of NaIO4 and NH2OH·HCl and 68% of desired product (2a) was observed. By increasing the proportion of NH2OH·HCl from 1 to 1.5, the desired product (2a) was increased to 87% yield. The maximum amount of yield was obtained by using 1:1.5 ratios of NaIO4 and NH2OH·HCl respectively. Further increasing the amount of both the reagents the yield of the product did not improve significantly.
:1 proportion of NaIO4 and NH2OH·HCl and 68% of desired product (2a) was observed. By increasing the proportion of NH2OH·HCl from 1 to 1.5, the desired product (2a) was increased to 87% yield. The maximum amount of yield was obtained by using 1:1.5 ratios of NaIO4 and NH2OH·HCl respectively. Further increasing the amount of both the reagents the yield of the product did not improve significantly.
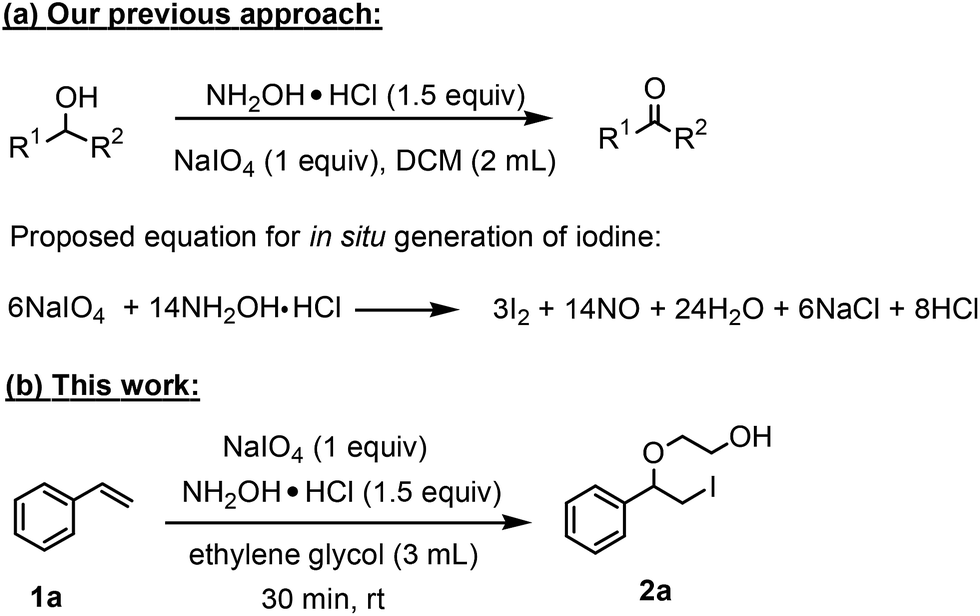
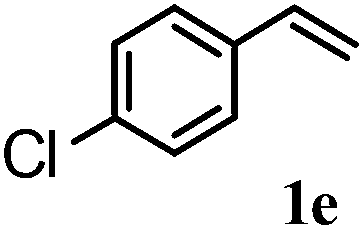
After optimizing the reaction conditions the scope and limitations of this reaction were investigated (Table 1). Our attention was focused on the use of different olefinic systems to prove the general applicability of the reaction conditions. It was observed that electron-rich and electron-deficient styrenes reacted efficiently with ethylene glycol to afford the desired products with good yields under the present reaction conditions. The styrene containing an electron donating Me & OMe group on the aromatic ring showed good efficiency (2b & 2c). The bromo- and chloro-substituted styrenes gave the corresponding 2d and 2e in 87% and 86% yields respectively without forming any dehalogenated products. Other electron withdrawing substituent NO2 group on styrene moiety afforded the desired product with satisfactory yield (2f). In addition, aliphatic olefinic systems were also found to afford the desired products with high yields (2g–2i). Our present protocol is also effective for cinnamyl alcohol to produce the corresponding β-iodo-β′-hydroxy ether (2j). We are pleased to notice that α-methyl styrene and 1,1-diphenylethylene both gave the desired products (2k & 2l) with good yields under the stated reaction conditions. However, sodium 4-vinyl benzenesulfonate, β-methyl-β-nitrostyrene, and cholesterol did not give the corresponding iodoethers under the present reaction conditions. This methodology is also applicable on a gram-scale synthesis. We have successfully prepared the iodoether 2a in 80% yield by the reaction of styrene (1a, 10 mmol) in ethylene glycol. In general, all the reactions were clean and β-iodo-β′-hydroxy ethers were found to be furnished regioselectively in all cases.
| Entry | Substrates (1) | Products (2) | Yieldsb (%) | |
|---|---|---|---|---|
| a All reactions were performed on a 1 mmol scale in presence of NaIO4 (1 mmol) and NH2OH·HCl (1.5 mmol) in 3 mL of ethylene glycol at room temperature for 30 min.b Isolated yields.c Styrene 1a (10 mmol), NaIO4 (10 mmol), NH2OH·HCl (15 mmol) in 30 mL of ethylene glycol at room temperature for 30 min.d cis product. | ||||
| 1 |  |
 |
2a | 87, 80c |
| 2 |  |
 |
2b | 83 |
| 3 |  |
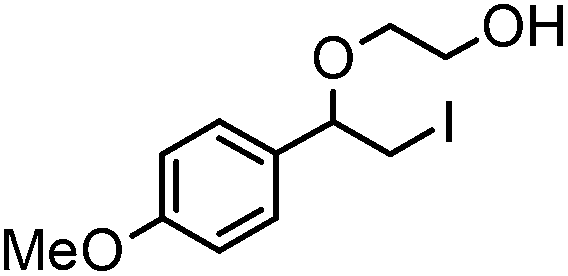 |
2c | 81 |
| 4 |  |
 |
2d | 87 |
| 5 | 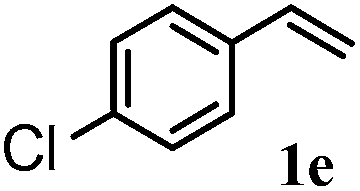 |
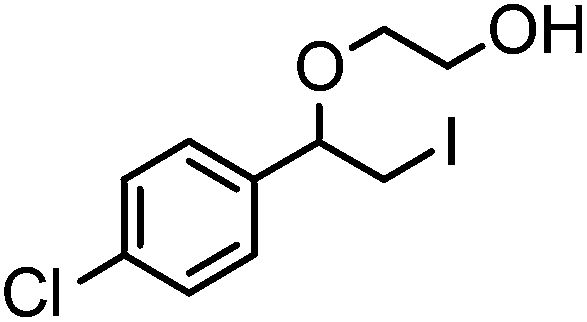 |
2e | 86 |
| 6 | 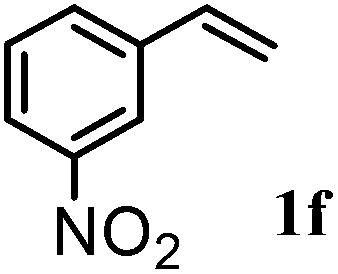 |
 |
2f | 81 |
| 7 |  |
 |
2g | 80 |
| 8 | 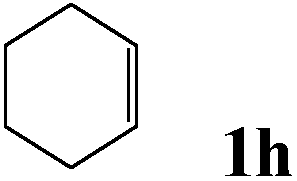 |
 |
2h | 82d |
| 9 |  |
 |
2i | 80d |
| 10 |  |
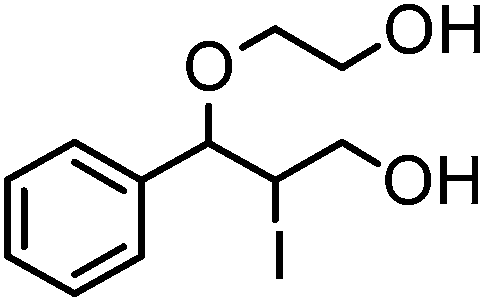 |
2j | 85 |
| 11 |  |
 |
2k | 82 |
| 12 |  |
 |
2l | 81 |
Next, we explored our present methodology using ethanol as other nucleophilic solvent to react with olefinic systems to synthesize various β-iodo ethers (Scheme 2). To our delight the corresponding β-iodo ethers (3) were obtained regioselectively in good yields; the results are summarized in Table 2.
 | ||
| Scheme 2 Synthesis of β-iodo ether using ethanol. | ||









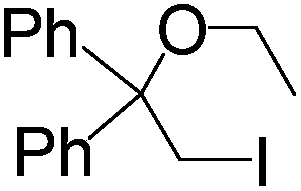


Simple styrene reacted well to give the desired β-iodo ether with high yield (3a). Styrenes substituted by electron donating OMe group (3b) as well as electron withdrawing halogen group (3c) underwent smooth reactions which highlighted the wide scope of this reaction. Meanwhile, the effect of alcoholic group in the olefinic system also investigated. Cinnamyl alcohol can also afford the desired product with excellent yield (3d). α-Methyl styrene and 1,1-diphenylethylene also nicely participated in the reaction, yielding the corresponding products 3e and 3f in 83% and 80% yields respectively. Moreover, aliphatic alkene such as 1-octene also afforded the desired product (3g) with good yield which also proves the general applicability of this present protocol.
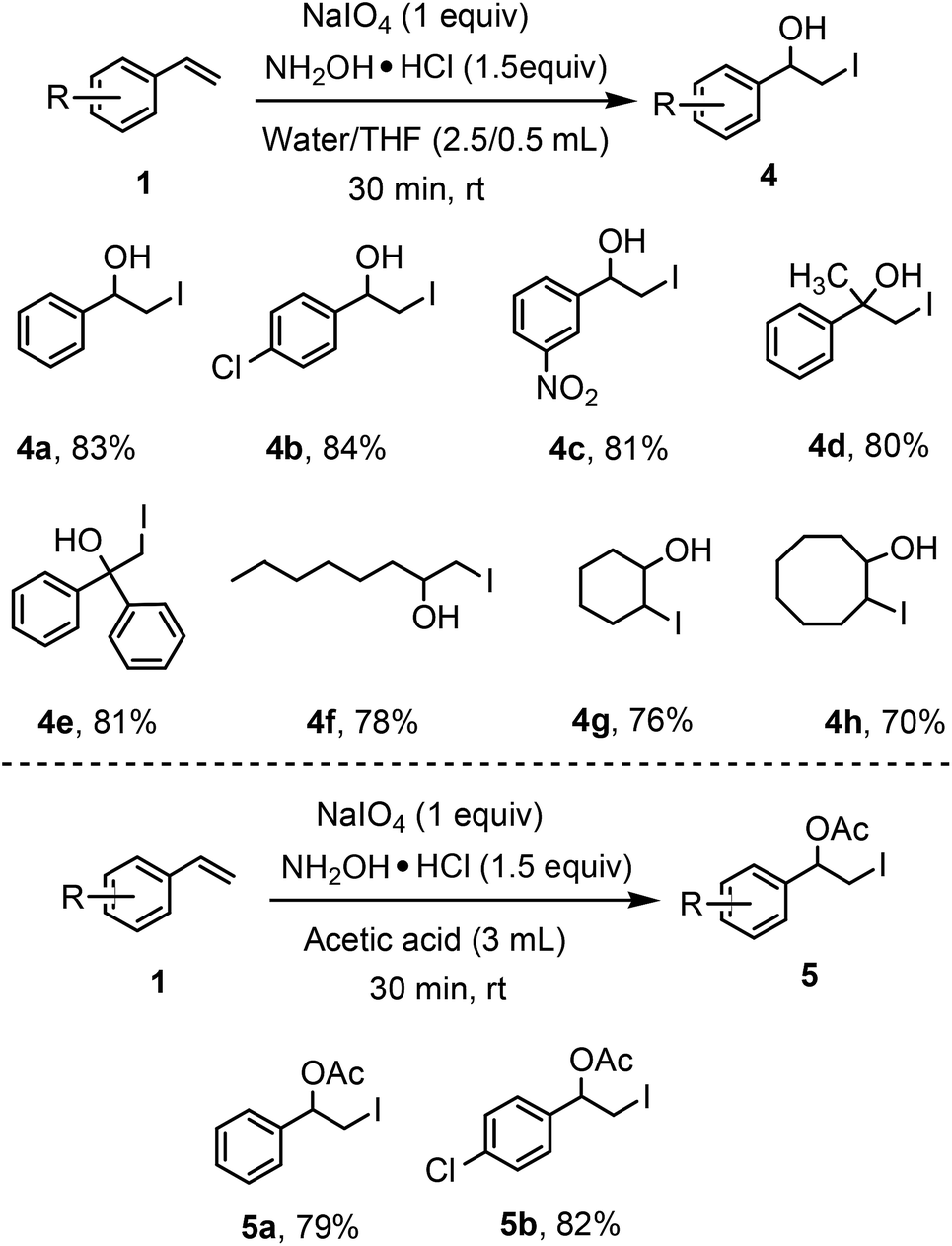
General applicability of the methodology has further been established by using the different solvents (nucleophiles) for the synthesis of iodohydrin and β-iodoacetoxy compounds (Scheme 3). We have successfully used water and acetic acid to synthesize iodohydrin (4) and β-iodoacetoxy compounds (5) with good yields which increases the scope of this transformation. It is worthy to mention that a little amount of THF was added to water as solvent to synthesize the iodohydrins. The styrene, substituted with chloro- and nitro-substituent smoothly afford the desired products (4a, 4b, 4c, 5a & 5b). We are delighted to inform that α-methyl styrene and 1,1-diphenylethylene both reacted well to give the corresponding iodohydrins (4d & 4e). Aliphatic olefins such as 1-octene, cyclohexene and cyclooctene can also afford the desired iodohydrins (4f–4h) with good yields. However, β-methyl-β-nitrostyrene did not give the corresponding iodohydrin under this present reaction conditions.
 | ||
| Scheme 3 Synthesis of iodohydrin and β-iodoacetoxy compounds. | ||
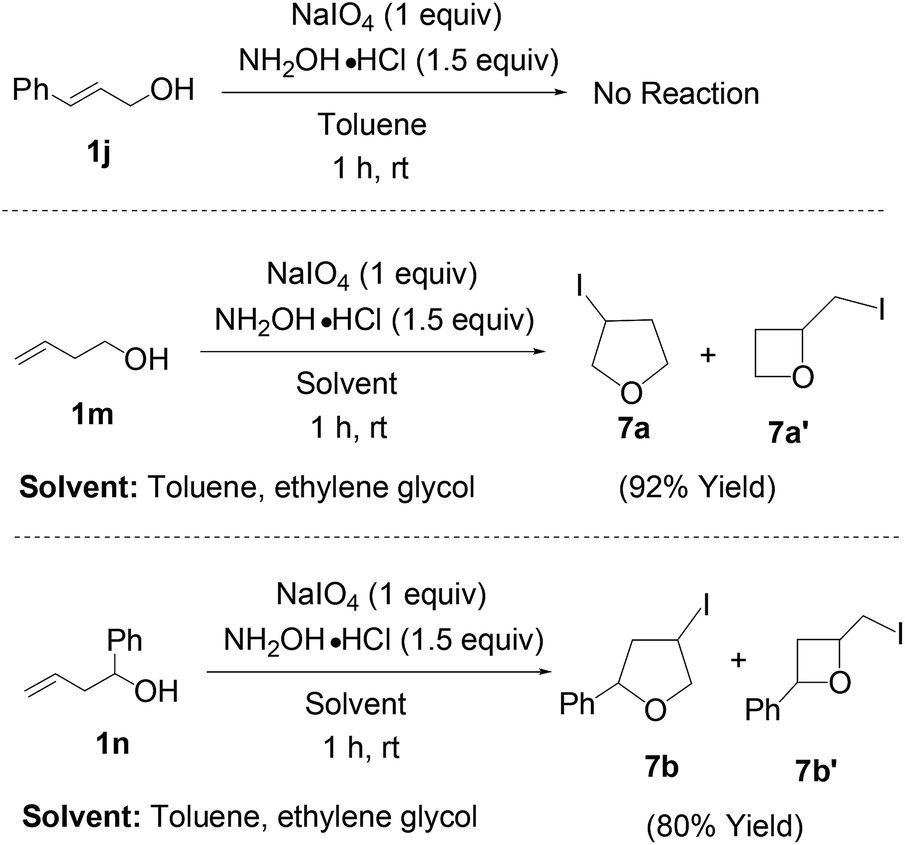
Next we have examined the possibility of intramolecular cyclization reaction under the present reaction conditions (Scheme 4). We have found that cinnamyl alcohol (1j) gave no reaction either oxidation31 to aldehyde or the intramolecular reaction by nucleophilic attack of alcoholic oxygen when the reaction has been carried out in non coordinating solvent like toluene. But it is worthy to mention that other homoallylic alcohols like 1m and 1n gave the cyclization products (7a, 7a′, 7b and 7b′) in presence of toluene or ethylene glycol. We have got inseparable mixture of products with good yields.
 | ||
| Scheme 4 Additional experiments on intramolecular cyclization reaction. | ||
Preparation of acetals at the terminal position of alkenes instead of aldehydes as substrate is a demanding task. Very few methods are available in literature using palladium,32 iron33 and a couple of nonmetal catalyzed methods.34 Recently, Narender et al. reported a metal-free approach for the synthesis of terminal acetals by tandem oxidative rearrangement of olefins using oxone as an oxidant in the presence of iodine.35 It is noteworthy to mention that our synthesized compound 2 is the key intermediate for synthesizing the terminal acetals. After successive reaction with β-iodo-β′-hydroxy ethers (2) using oxone as oxidant, various terminal acetals (6a–6j) were successfully synthesized by employing the reported method (Scheme 5).35
 | ||
| Scheme 5 Synthesis of terminal acetals from β-iodo-β′-hydroxy ethers. | ||
Conclusions
In summary, we have developed a simple and general method for the synthesis of β-iodo-β′-hydroxy ether, β-iodo ether, β-iodo hydrin, and β-iodo acetoxy compounds using the combination of NH2OH·HCl and NaIO4 as iodine source at room temperature within a short reaction time using different solvents which act as nucleophiles. Furthermore, the β-iodo-β′-hydroxy ethers have been converted to terminal acetals using the reported method by using oxone as oxidant. The preparation of terminal acetals using β-iodo-β′-hydroxy ether is the very important functionalization of alkene as we can functionalize the germinal position by non-Wacker reaction. The advantages of this present protocol are: (i) molecular iodine-free synthesis, (ii) room temperature and mild reaction conditions, (iii) short reaction time, (iv) high yields, (v) use of less expensive reagents, (vi) applicable for broad solvent (nucleophile) systems, and (vii) applicable for gram-scale synthesis. These advantages render this protocol facile and suitable to create a diversified library of β-iodo-β′-hydroxy ether, β-iodo ether, β-iodo hydrin, and β-iodo acetoxy compounds.Experimental section
General experimental methods
1H NMR spectra were determined on a Bruker 400 (400 MHz) spectrometer as solutions in CDCl3. Chemical shifts are expressed in parts per million (δ) and are referenced to tetramethylsilane (TMS) as internal standard and the signals were reported as s (singlet), d (doublet), t (triplet), m (multiplet) and coupling constants J were given in Hz. 13C NMR spectra were recorded at 100 MHz in CDCl3 solution. TLC was done on silica gel coated glass slide (Merck, Silica gel G for TLC). Silica gel (60–120 mesh, SRL, India) was used for column chromatography. Petroleum ether refers to the fraction boiling in the range of 60–80 °C unless otherwise mentioned. Melting points were determined on a glass disk with an electric hot plate and are uncorrected. All solvents were dried and distilled before use. Commercially available substrates were freshly distilled before the reaction. Solvents, reagents and chemicals were purchased from Aldrich, Fluka, Merck, SRL, Spectrochem and Process Chemicals. All reactions involving moisture sensitive reactants were executed using oven dried glassware.Typical procedure for the synthesis of compound 2
A mixture of alkene (1 mmol), NaIO4 (1 mmol, 213 mg) in 3 mL of ethylene glycol was taken in a round bottomed flask at room temperature and then NH2OH·HCl (1.5 mmol, 104 mg) was added by portion for 5 min. The reaction mixture was stirred for 30 min at room temperature. After completion (TLC), the reaction mixture was diluted with a 1:1 mixture of water/ethyl acetate (10 mL) and washed with 10% (w/v) Na2S2O3 (3 × 5 mL) followed by brine solution (1 × 10 mL). Then the combined organic layer was dried over anhydrous Na2SO4. Evaporation of solvent furnished the crude product which was subjected to column chromatography using ethyl acetate–petroleum ether (1:15) as eluent to obtain the analytically pure product.
Typical procedure for the synthesis of compound 2a on gram-scale
A mixture of styrene 1a (10 mmol, 1.04 g), NaIO4 (10 mmol, 2.13 g) in 30 mL of ethylene glycol was taken in a round bottomed flask at room temperature and then NH2OH·HCl (15 mmol, 1.04 g) was added by portion for 5–10 min. The reaction mixture was stirred for 30 min at room temperature. After completion (TLC), the reaction mixture was diluted with a 1:1 mixture of water/ethyl acetate (50 mL) and washed with 10% (w/v) Na2S2O3 (3 × 20 mL) followed by brine solution (1 × 30 mL). Then the combined organic layer was dried over anhydrous Na2SO4. Evaporation of solvent furnished the crude product which was subjected to column chromatography using ethyl acetate–petroleum ether (1:15) as eluent to obtain the analytically pure product.
Typical procedure for the synthesis of compound 3
A mixture of alkene (1 mmol), NaIO4 (1 mmol, 213 mg) in 3 mL of ethanol was taken in a round bottomed flask at room temperature and then NH2OH·HCl (1.5 mmol, 104 mg) was added by portion for 5 min. The reaction mixture was stirred for 30 min at room temperature. After completion (TLC), the reaction mixture was diluted with a 1:1 mixture of water/ethyl acetate (10 mL) and washed with 10% (w/v) Na2S2O3 (3 × 5 mL) followed by brine solution (1 × 10 mL). Then the combined organic layer was dried over anhydrous Na2SO4. Evaporation of solvent furnished the crude product which was subjected to column chromatography using ethyl acetate–petroleum ether (1:15) as eluent to obtain the analytically pure product.
Typical procedure for the synthesis of compound 4
A mixture of alkene (1 mmol), NaIO4 (1 mmol, 213 mg) in a mixture of 2.5 mL of water and 0.5 mL of THF was taken in a round bottomed flask at room temperature and then NH2OH·HCl (1.5 mmol, 104 mg) was added by portion for 5 min. The reaction mixture was stirred for 30 min at room temperature. After completion (TLC), the reaction mixture was diluted with a 1:1 mixture of water/ethyl acetate (10 mL) and washed with 10% (w/v) Na2S2O3 (3 × 5 mL) followed by brine solution (1 × 10 mL). Then the combined organic layer was dried over anhydrous Na2SO4. Evaporation of solvent furnished the crude product which was subjected to column chromatography using ethyl acetate–petroleum ether (1:15) as eluent to obtain the analytically pure product.
Typical procedure for the synthesis of compound 5
A mixture of alkene (1 mmol), NaIO4 (1 mmol, 213 mg) in 3 mL of acetic acid was taken in a round bottomed flask at room temperature and then NH2OH·HCl (1.5 mmol, 104 mg) was added by portion for 5 min. The reaction mixture was stirred for 30 min at room temperature. After completion (TLC), the reaction mixture was diluted with a 1:1 mixture of water/ethyl acetate (10 mL) and washed with 10% (w/v) Na2S2O3 (3 × 5 mL) followed by brine solution (1 × 10 mL). Then the combined organic layer was dried over anhydrous Na2SO4. Evaporation of solvent furnished the crude product which was subjected to column chromatography using ethyl acetate–petroleum ether (1:15) as eluent to obtain the analytically pure product.
Typical procedure for the synthesis of compound 6 (ref. 35)
Oxone (0.75 mmol) was slowly added to compound 2 (1 mmol) in 2 mL of ethylene glycol in a round bottomed flask and the reaction mixture was stirred at room temperature for 2 h. After completion (TLC), the reaction mixture was diluted with a 1:1 mixture of water/DCM (10 mL) and washed with 10% (w/v) Na2S2O3 (2 × 5 mL) followed by brine solution (1 × 10 mL). Then the combined organic layer was dried over anhydrous Na2SO4. Evaporation of solvent furnished the crude product which was subjected to column chromatography using ethyl acetate–petroleum ether (1:15) as eluent to obtain the analytically pure product.
Typical procedure for the synthesis of compound 7
A mixture of alkene (1 mmol), NaIO4 (1 mmol, 213 mg) in 3 mL of toluene was taken in a round bottomed flask at room temperature and then NH2OH·HCl (1.5 mmol, 104 mg) was added by portion for 5 min. The reaction mixture was stirred for 1 h at room temperature. After completion (TLC), the reaction mixture was diluted with a 1:1 mixture of water/ethyl acetate (10 mL) and washed with 10% (w/v) Na2S2O3 (3 × 5 mL) followed by brine solution (1 × 10 mL). Then the combined organic layer was dried over anhydrous Na2SO4. Evaporation of solvent furnished the crude product which was subjected to column chromatography using ethyl acetate–petroleum ether (1:15) as eluent to obtain the product.
Acknowledgements
A. Majee is pleased to acknowledge the financial support from BRNS, Govt. of India (Grant No. 37(2)/14/35/2014-BRNS/563). A. Hajra acknowledges CSIR, New Delhi, India (Grant No. 02(0168)/13/EMR-II) for financial support. We are thankful to DST-FIST and UGC-SAP. S. Santra and G. V. Zyryanov acknowledge the Russian Scientific Fund (Grant # 15-13-10033) for funding.Notes and references
- (a) E. Block and A. L. Schwan, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1st edn, 1991, vol. 4, p. 329 Search PubMed; For reviews see: (b) A. Minatti and K. Muñiz, Chem. Soc. Rev., 2007, 36, 1142 RSC; (c) K. H. Jensen and M. S. Sigman, Org. Biomol. Chem., 2008, 6, 4083 RSC; (d) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS PubMed; (e) D. M. Schultz and J. P. Wolfe, Synthesis, 2012, 351 CAS.
- (a) R. E. Erickson, in Marine Natural Products, ed. P. J. Scheuer, Academic Press, New York, 1986, vol. 5, p. 131 Search PubMed; (b) P. A. Bartlett, in Asymmetric Synthesis, ed. J. D. Morrison, Academic Press, New York, 1984, vol. 3, p. 411 Search PubMed; (c) I. Cabanal-Duvillard, J.-F. Berrier, J. Royer and H.-P. Husson, Tetrahedron Lett., 1998, 39, 5181 CrossRef CAS; (d) J. P. Konopelski, M. A. Boehler and T. M. Tarasow, J. Org. Chem., 1989, 54, 4966 CrossRef CAS; (e) Y. Ueda and S. C. Maynard, Tetrahedron Lett., 1988, 29, 5197 CrossRef CAS; (f) C. Christophersen, Acta Chem. Scand., 1985, 39B, 517 CrossRef PubMed.
- P. B. D. de la Mare, Electrophilic Halogenation, Cambridge University Press, London, 1976 Search PubMed.
- A. M. Sanseverino, F. M. da Silva, J. Jones Jr and M. C. S. de Mattos, Quim. Nova, 2001, 24, 637 CrossRef CAS PubMed.
- (a) J. Rodriguez and J. P. Dulcere, Synthesis, 1993, 1177 CrossRef CAS PubMed, and references cited therein. (b) G. K. Dewkar, S. V. Narina and A. Sudalai, Org. Lett., 2003, 5, 4501 CrossRef CAS PubMed.
- K. Weissermel, Industrial Organic Chemistry, Wiley-VCH, Wienheim, 1997, p. 266 Search PubMed.
- J. W. Cornforth and D. T. Green, J. Chem. Soc. C, 1970, 846 RSC.
- (a) J. Bougault, C. R. Acad. Sci., 1900, 130, 1766 CAS; (b) J. Bougault, C. R. Acad. Sci., 1900, 131, 528 CAS.
- J. Barluenga, M. A. Rodriguez, P. J. Campos and G. Asensio, J. Chem. Soc., Chem. Commun., 1987, 1491 RSC.
- (a) M. Parrilli, G. Barone, M. Adinolfi and L. Mangoni, Tetrahedron Lett., 1976, 17, 207 CrossRef; (b) R. Antonioletti, M. D'Auria, A. De Mico, G. Piancatelli and A. Scettri, Tetrahedron, 1983, 39, 1765 CrossRef CAS.
- J. G. Smith and M. Fieser, in Fieser and Fieser's Reagent for Organic Synthesis, John Wiley and Sons, New York, vol. 1–12, 1990 Search PubMed.
- (a) J. G. Smith, Synthesis, 1984, 629 CrossRef CAS; (b) A. S. Rao, S. K. Paknikar and J. G. Kirtane, Tetrahedron, 1983, 39, 2323 CrossRef CAS.
- (a) S. Y. Liu and D. G. Nocera, Tetrahedron Lett., 2006, 47, 1923 CrossRef CAS PubMed; (b) R. I. Kureshy, S. Singh, N. H. Khan, S. H. R. Abdi, I. Ahmed, A. Bhatt and R. V. Jasra, Catal. Lett., 2006, 107, 127 CrossRef CAS; (c) S. L. H. Rebelo, A. R. Gonçalves, M. M. Pereira, M. M. Q. Simões, M. G. P. M. S. Neves and J. A. S. Cavaleiro, J. Mol. Catal. A: Chem., 2006, 256, 321 CrossRef CAS PubMed.
- A. Corma, J. Catal., 2003, 216, 298 CrossRef CAS.
- G. Grigoropoulou, J. H. Clark and J. A. Elings, Green Chem., 2003, 5, 1 RSC.
- (a) M. Fieser and L. F. Fieser, in Reagents for Organic Synthesis, ed. J. G. Smith and M. Fieser, John Wiley and Sons, New York, 1990 Search PubMed; (b) G. Majetich, R. Hicks and S. Reister, J. Org. Chem., 1997, 62, 4321 CrossRef CAS PubMed; (c) N. Iranpoor and M. Shekarriz, Tetrahedron, 2000, 56, 5209 CrossRef CAS; (d) J. Barluenga, M. Marco-Arias, F. Gonzáles-Bobes, A. Ballesteros and J. M. Gonzáles, Chem.–Eur. J., 2004, 10, 1677 CrossRef CAS PubMed; (e) B. C. Ranu and S. Banerjee, J. Org. Chem., 2005, 70, 4517 CrossRef CAS PubMed; (f) B. Das, K. Venkateswarlu, K. Damodar and K. Suneel, J. Mol. Catal. A: Chem., 2007, 269, 17 CrossRef CAS PubMed; (g) Shallu, M. L. Sharma and J. Singh, Synth. Commun., 2012, 42, 1306 CrossRef CAS PubMed.
- Y. Guindon, B. Guérin, C. Chabot, H. Mackintosh and W. W. Olgivie, Synlett, 1995, 449 CrossRef CAS PubMed.
- K. Maeda, H. Shinokubo and K. Oshima, J. Org. Chem., 1996, 61, 6770 CrossRef CAS.
- J. Barluenga, J. M. González, P. J. Campos and G. A. Asensio, Angew. Chem., Int. Ed., 1985, 24, 319 CrossRef PubMed.
- A. M. Sanseverino and M. C. S. de Mattos, Synthesis, 1998, 1584 CrossRef CAS PubMed.
- A. Kirschning, E. Kunst, M. Ries, L. Rose, A. Schönberger and R. Wartchow, ARKIVOC, 2003, 145 CrossRef CAS.
- R. A. S. Villegas, J. L. E. Santo Jr, M. C. S. de Mattos, M. R. M. P. de Aguiar and A. W. S. Guarino, J. Braz. Chem. Soc., 2005, 16, 565 CrossRef CAS PubMed.
- (a) K. Rama and M. A. Pasha, Ultrason. Sonochem., 2005, 12, 437 CrossRef CAS PubMed; (b) V. S. Fernandes, J. C. S. Barboza and A. A. Serra, Synth. Commun., 2007, 37, 1433 CrossRef CAS PubMed.
- E. Kolvani, A. Ghorbani-Choghamarani, P. Salehi, F. Shirini and M. A. Zolfigol, J. Iran. Chem. Soc., 2007, 4, 126 CrossRef.
- S. P. L. De Souza, J. F. M. da Silva and M. C. S. de Mattos, Quim. Nova, 2006, 29, 1061 CrossRef CAS PubMed.
- (a) M. Smietana, V. Gouverneur and C. Mioskowski, Tetrahedron Lett., 2000, 41, 193 CrossRef CAS; (b) D. S. Middleton and N. S. Simpkins, Synth. Commun., 1989, 19, 21 CrossRef CAS PubMed.
- R. D. S. Ribeiro, P. M. Esteves and M. C. S. de Mattos, Tetrahedron Lett., 2007, 48, 8747 CrossRef CAS PubMed.
- J. N. Moorthy, K. Senapati and S. Kumar, J. Org. Chem., 2009, 74, 6287 CrossRef CAS PubMed.
- H. Togo and S. Iida, Synlett, 2006, 2159 CrossRef CAS PubMed.
- (a) S. Mitra, A. Chakraborty, S. Mishra, A. Majee and A. Hajra, Org. Lett., 2014, 16, 5652 CrossRef CAS PubMed; (b) S. Santra, S. Mitra, A. K. Bagdi, A. Majee and A. Hajra, Tetrahedron Lett., 2014, 55, 5151 CrossRef CAS PubMed; (c) K. Monir, M. Ghosh, S. Mishra, A. Majee and A. Hajra, Eur. J. Org. Chem., 2014, 1096 CrossRef CAS PubMed; (d) K. Monir, A. K. Bagdi, S. Mishra, A. Majee and A. Hajra, Adv. Synth. Catal., 2014, 356, 1105 CrossRef CAS PubMed; (e) S. Santra, A. K. Bagdi, A. Majee and A. Hajra, Adv. Synth. Catal., 2013, 355, 1065 CrossRef CAS PubMed; (f) A. K. Bagdi, M. Rahman, S. Santra, A. Majee and A. Hajra, Adv. Synth. Catal., 2013, 355, 1741 CrossRef CAS PubMed.
- A. Majee, S. K. Kundu, S. Santra and A. Hajra, Tetrahedron Lett., 2012, 53, 4433 CrossRef CAS PubMed.
- (a) M. Yamamoto, S. Nakaoka, Y. Ura and Y. Kataoka, Chem. Commun., 2012, 48, 1165 RSC; (b) A. Kishi, S. Sakaguchi and Y. Ishii, Org. Lett., 2000, 2, 523 CrossRef CAS PubMed; (c) T. Hosokawa, T. Ohta and S.-I. Murahashi, J. Chem. Soc., Chem. Commun., 1983, 848 RSC; (d) T. Hosokawa, T. Ohta, S. Kanayama and S.-I. Murahashi, J. Org. Chem., 1987, 52, 1758 CrossRef CAS; (e) F. Alonso, D. Sanchez, T. Soler and M. Yus, Adv. Synth. Catal., 2008, 350, 2118 CrossRef CAS PubMed; (f) P. M. Tadross, P. Bugga and B. M. Stoltz, Org. Biomol. Chem., 2011, 9, 5354 RSC.
- A. D. Chowdhury and G. K. Lahiri, Chem. Commun., 2012, 48, 3448 RSC.
- (a) M. Ochiai, K. Miyamoto, M. Shiro, T. Ozawa and K. Yamaguchi, J. Am. Chem. Soc., 2003, 125, 13006 CrossRef CAS PubMed; (b) M. S. Yusubov and G. A. Zholobova, Russ. J. Org. Chem., 2001, 37, 1179 CrossRef CAS.
- M. A. Kumar, P. Swamy, M. Naresh, M. M. Reddy, C. N. Rohitha, S. Prabhakar, A. V. S. Sarma, J. R. P. Kumar and N. Narender, Chem. Commun., 2013, 49, 1711 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for all compounds. See DOI: 10.1039/c5ra11092k |
| This journal is © The Royal Society of Chemistry 2015 |