
Carbocationic polymerization of isoprene using cumyl initiators: progress in understanding side reactions†
Abstract
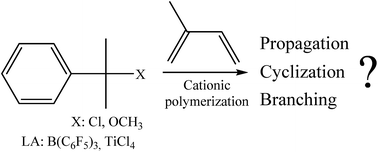
The cationic polymerization of isoprene using cumyl chloride/B(C6F5)3 and cumyl ether/TiCl4 systems was investigated in dichloromethane or in dichloromethane/methylcyclohexane mixtures varying the polymerization conditions. Polymerizations were performed in all cases in the presence of a large excess of a proton trap (2,6-di-tert-butyl pyridine, dtBP) compared to initiator in order to suppress any protic side reactions. As a consequence, no polymerization went to completion. Independently of the reaction conditions, trans-1,4-oligomers were exclusively obtained with mainly an olefinic terminal group. It was highlighted that an important loss of double bonds yielding saturated parts was observed, even in the absence of protons, assuming that a great amount of double bond loss generally observed in isoprene cationic polymerization could be due to intramolecular cyclization reactions. Nevertheless, under particular conditions (low temperature and/or low polarity medium), branching and cross-linking reactions were also found responsible for double bond loss.
 Please wait while we load your content...
Please wait while we load your content...

